Introduction
(Article introduction authored by Conquest Editorial Team)
Over the past 50 years, aplastic anemia (AA) treatment has transformed from a fatal diagnosis to one with survival rates exceeding 80–85%. Hematopoietic stem cell transplantation (HSCT) and immunosuppressive therapy (IST) are now the primary treatments, with IST being more widely used due to donor scarcity, patient age, comorbidities, and limited HSCT access. A key breakthrough was the introduction of antithymocyte globulin (ATG), which, when combined with cyclosporine, significantly improved treatment outcomes.
In the late 1980s, combining ATG with cyclosporine became the standard treatment for severe AA (SAA), achieving a 70% response rate and improved survival compared to ATG alone. The most effective current regimen for SAA includes horse ATG, cyclosporine, and eltrombopag, which represents a major advancement in AA management. This progress marks a significant improvement in outcomes for patients with this once-lethal disease.
Aplastic anemia as an immune disorder
Aplastic anemia is characterized by pancytopenia and a hypocellular marrow, diagnosed after excluding other causes. In young patients, it’s crucial to rule out inherited marrow failure syndromes like Fanconi anemia, telomeropathies, and ribosomopathies, while in older patients, it must be differentiated from myelodysplastic syndromes (MDS). Severe aplastic anemia is diagnosed when at least two of these criteria are met: neutrophil count below 500/µL, reticulocyte count under 60,000/µL, or platelet count less than 20,000/µL.
Genetic testing for mutations like GATA2 and RUNX1 is important, especially in younger patients. Marrow flow cytometry and cytogenetics help distinguish aplastic anemia from hypoplastic MDS in older patients.
The presence of a PNH clone suggests an immune etiology, and autoimmune origins of the disease are often indicated by circumstantial evidence, such as lymphocyte infiltration or positive responses to immunosuppressive therapy. (Fig. 1)

In aplastic anemia (AA), immune system abnormalities lead to the destruction of marrow progenitor cells, driven by elevated inflammatory cytokines like interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α). This destruction occurs via a Fas-dependent pathway. CD8+ cell expansion with shared T-cell receptors suggests antigen-driven immune skewing, though the specific antigen is unknown.
Genetic loss of one HLA haplotype on myeloid cells may allow these cells to escape immune destruction, indicating immune pressure on hematopoietic stem cells (HSCs). Reduced regulatory T cells (Tregs) at diagnosis result in unchecked inflammation, reflected in a high Th17/Treg ratio. Experimental models confirm the immune system’s role in AA, which often responds to immunosuppressive therapy.
Therapy
The current treatment paradigm relies on age and availability of a histocompatible donor. For decades, efforts to improve severe aplastic anemia (SAA) treatment focused on intensifying immunosuppression, but adding third immunosuppressants, androgens, or growth factors to the standard h-ATG/CsA regimen did not yield better outcomes.
More potent agents like alemtuzumab and rabbit ATG (r-ATG) also failed to enhance recovery, with h-ATG consistently proving superior. The exploration of novel IST regimens dwindled until thrombopoietin receptor agonists (Tpo-RAs), initially developed for immune thrombocytopenia, were tested in SAA. Eltrombopag showed promise in refractory cases, with a 50% hematologic response and multilineage improvements in some patients.
It was later tested as a front-line therapy combined with h-ATG/CsA (ACE regimen), showing significant improvement in complete response rates and overall survival. The European Society for Blood and Marrow Transplantation’s RACE trial confirmed the ACE regimen as the new standard for first-line therapy in AA, demonstrating superior outcomes compared to the previous standard.
Why did it take so long?
Despite extensive research from murine models, most AA treatment protocols emerged from insights in other medical fields. The use of ATG in AA was discovered by chance, and cyclosporine was added in the 1980s based on its success in treating T-cell-mediated disorders.
Over 30 years, treatment strategies focused on intensifying IST, but attempts to stimulate progenitor cells with growth factors were largely unsuccessful. Thrombopoietin receptor agonists (Tpo-RAs) were explored due to their role in early progenitor cells, despite skepticism from thrombopoietin levels in AA patients.
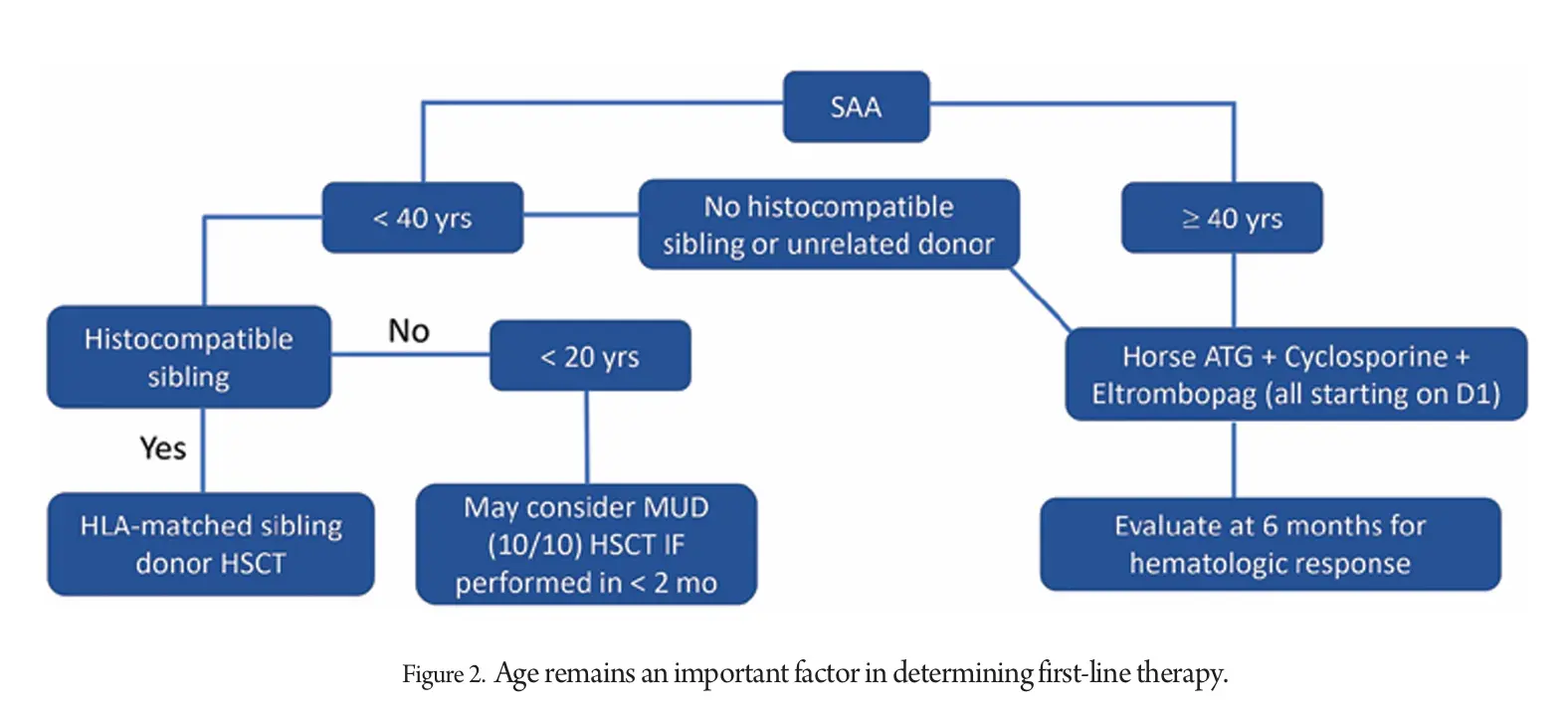
Trials aimed at intensifying IST yielded disappointing results, suggesting a potential threshold of effectiveness. Combining h-ATG and cyclosporine with Tpo-RAs like eltrombopag led to unexpected success, with significant hematologic recoveries and the possibility of discontinuing treatment in some patients.
Eltrombopag’s success may be due to its immunomodulatory effects, promotion of TGF-β secretion, reduction of pro-inflammatory cytokines, iron chelation, and counteracting IFNγ’s impact on marrow cells. The positive results with romiplostim suggest a class effect among Tpo-RAs in AA treatment.
Where do we go from here?
Currently, the combination of h-ATG, cyclosporine, and eltrombopag is the most effective IST regimen for severe aplastic anemia (SAA), especially in adults, showing less benefit in children due to their higher number of HSCs and better response to h-ATG plus cyclosporine alone. For patients not responding to this regimen, HSCT is an option, while those ineligible may consider repeated immunosuppression, supportive care, growth factors, and androgens.
Despite advancements, treatment decisions still hinge on age and donor availability. Telomere biology, although explored, has not yet influenced treatment algorithms due to variable gene mutation penetrance.
New treatments are being developed, including JAK pathway inhibitors, regulatory T cell promoters, and IFNγ inhibitors, along with alternative TPO-RAs. Combining androgens or growth factors with the current regimen holds potential, and further progress in AA treatment is eagerly anticipated.
References
1. Kojima S, Horibe K, Inaba J, Yoshimi A, Takahashi Y, Kudo K, et al. Long-term outcome of acquired aplastic anaemia in children: comparison between immunosuppressive therapy and bone marrow transplantation. Br J Haematol. 2000;111:321–8.
2. Yoshida N, Kobayashi R, Yabe H, Kosaka Y, Yagasaki H, Watanabe K, et al. First-line treatment for severe aplastic anemia in children: bone marrow transplantation from a matched family donor versus immunosuppressive therapy. Haematologica. 2014;99:1784–91.
3. Dufour C, Pillon M, Socie G, Rovo A, Carraro E, Bacigalupo A, et al. Outcome of aplastic anaemia in children. A study by the severe aplastic anaemia and paediatric disease working parties of the European group blood and bone marrow transplant. Br J Haematol. 2015;169:565–73.
4. Atmar K, Ruivenkamp CAL, Hooimeijer L, Nibbeling EAR, Eckhardt CL, Huisman EJ, et al. Diagnostic value of a protocolized in-depth evaluation of pediatric bone marrow failure: a multi-center prospective cohort study. Front Immunol. 2022;13: 883826.
5. Keel S, Geddis A. The clinical and laboratory evaluation of patients with suspected hypocellular marrow failure. Hematol Am Soc Hematol Educ Program. 2021;1:134–42.
6. Shimano KA, Narla A, Rose MJ, Gloude NJ, Allen SW, Bergstrom K, et al. Diagnostic work-up for severe aplastic anemia in children: consensus of the North American pediatric aplastic anemia consortium. Am J Hematol. 2021;96:1491–504.
7. Muramatsu H, Okuno Y, Yoshida K, Shiraishi Y, Doisaki S, Narita A, et al. Clinical utility of next-generation sequencing for inherited bone marrow failure syndromes. Genet Med. 2017;19:796–802.
8. Ohara A, Kojima S, Hamajima N, Tsuchida M, Imashuku S, Ohta S, et al. Myelodysplastic syndrome and acute myelogenous leukemia as a late clonal complication in children with acquired aplastic anemia. Blood. 1997;90:1009–13.
9. Socie G, Henry-Amar M, Bacigalupo A, Hows J, Tichelli A, Ljungman P, et al. Malignant tumors occurring after treatment of aplastic anemia. European bone marrow transplantation-severe aplastic anaemia working party. N Engl J Med. 1993;329:1152–7.
10. Yoshida N, Yagasaki H, Hama A, Takahashi Y, Kosaka Y, Kobayashi R, et al. Predicting response to immunosuppressive therapy in childhood aplastic anemia. Haematologica. 2011;96:771–4.
11. Narita A, Zhu X, Muramatsu H, Chen X, Guo Y, Yang W, et al. Prospective randomized trial comparing two doses of rabbit anti-thymocyte globulin in patients with severe aplastic anaemia. Br J Haematol. 2019;187:227–37.
12. Kikuchi A, Yabe H, Kato K, Koh K, Inagaki J, Sasahara Y, et al. Long-term outcome of childhood aplastic anemia patients who underwent allogeneic hematopoietic SCT from an HLA-matched sibling donor in Japan. Bone Marrow Transplant. 2013;48:657–60.
13. Vo P, Onstad L, Flowers ME, Storb R. Cancers after HLA-matched related bone marrow transplantation for aplastic anemia. Bone Marrow Transplant. 2022;57:83–8.