
Abstract
In the treatment of rheumatoid arthritis (RA), Janus kinase inhibitors (jakinibs) represent an emerging class of targeted therapies in addition to biologics. The number of jakinibs has been growing and as of 2020, filgotinib was the latest jakinib to enter the international market for treating RA. Filgotinib has demonstrated preferential inhibition of JAK1-dependent cytokine signaling in in vitro assays. It has been evaluated in the DARWIN (phase 2) and FINCH (phase 3) series of clinical studies for treating patients with moderately-to-severely active RA. Filgotinib received regulatory approval in Japan and Europe in September 2020, while in August 2020 the United States Food and Drug Administration requested additional data from two ongoing clinical studies assessing the potential impact of filgotinib on sperm parameters. This article will review the pharmacological properties, efficacy, and safety of filgotinib as demonstrated in clinical studies. Expert opinion will be provided on jakinibs for RA treatment from the viewpoints of basic research and clinical practice.
Introduction
Rheumatoid arthritis (RA) is a chronic, immune-mediated inflammatory disease (IMID) characterized by swollen, painful joints and the presence of autoimmunity markers. The current model of the pathogenesis of RA suggests that autoimmunity and inflammation are triggered at mucosal sites initially, followed by their systemic expansion and the development of clinical symptoms [1]. It remains unclear in what sequence early inflammation and autoimmunity develop in the pre-RA phase, or if specific inflammatory pathways are causing the development of systemic autoimmunity [1]. Abnormalities of numerous cytokines involved in immune and inflammation responses have been implicated in RA [2]. The spectrum of signaling pathways responsible for RA pathogenesis remains incompletely understood.
A recent development in the treatment of RA has been the advent of Janus kinase inhibitors (jakinibs), a new class of disease-modifying anti-rheumatic drugs (DMARDs). Jakinibs are small-molecule, targeted therapies that inhibit the JAK signaling-dependent enzymes and, in turn, block the intracellular signal transduction downstream of a number of cytokine and growth factor receptor stimulation [3,4]. Jakinibs offer potentially rapid onset of efficacy, oral administration, and absence of immunogenicity. Disadvantages, however, remain an increased risk of infection and the need for laboratory monitoring. Compared with monoclonal antibodies, small-molecule inhibitors may be more prone to having off-target side effects. The long-term safety data currently available for jakinibs are also less than those for biological and conventional synthetic DMARDs (bDMARDs and csDMARDs). In view of emerging data, the EULAR RA management guidelines recently (late 2019) raised the level of recommendation for jakinibs to be equal to that for bDMARDs as second- and third-line treatment options, to be used in combination with csDMARDs in patients with inadequate responses (IRs) to earlier phases of treatment as guided by the treatment-to-target principle [5].
The JAK family of enzymes consists of four members, namely, JAK1, JAK2, JAK3, and TYK2. These JAK isoforms form homo- and hetero-dimers and one heterotrimer that facilitate the signal transduction of various type I/II cytokines and growth factors implicated in inflammatory and hematopoietic pathways (Figure 1) [6,7]. Remarkable signaling versatility and complexity arise from the array of JAK dimers, as well as the numerous ‘cytokine–JAK dimer–signal transducer and activator of transcription (STAT) effector’ combinations, which present both opportunities and challenges in the endeavor of treating RA through JAK inhibition [6,8]. For example, JAK1 inhibition may simultaneously block the signaling of IL-6 and IFNα, both of which are implicated in RA pathology. On the other hand, inhibiting JAK2 may hamper JAK2/2 homodimer-dependent processes such as erythropoiesis (and, possibly, JAK2-containing heterodimer-mediated immune responses) (Figure 1), potentially leading to undesirable side effects. As such, the selectivity profile of jakinibs and its potential relationship to jakinibs’ clinical efficacy and safety have been receiving increased research attention.
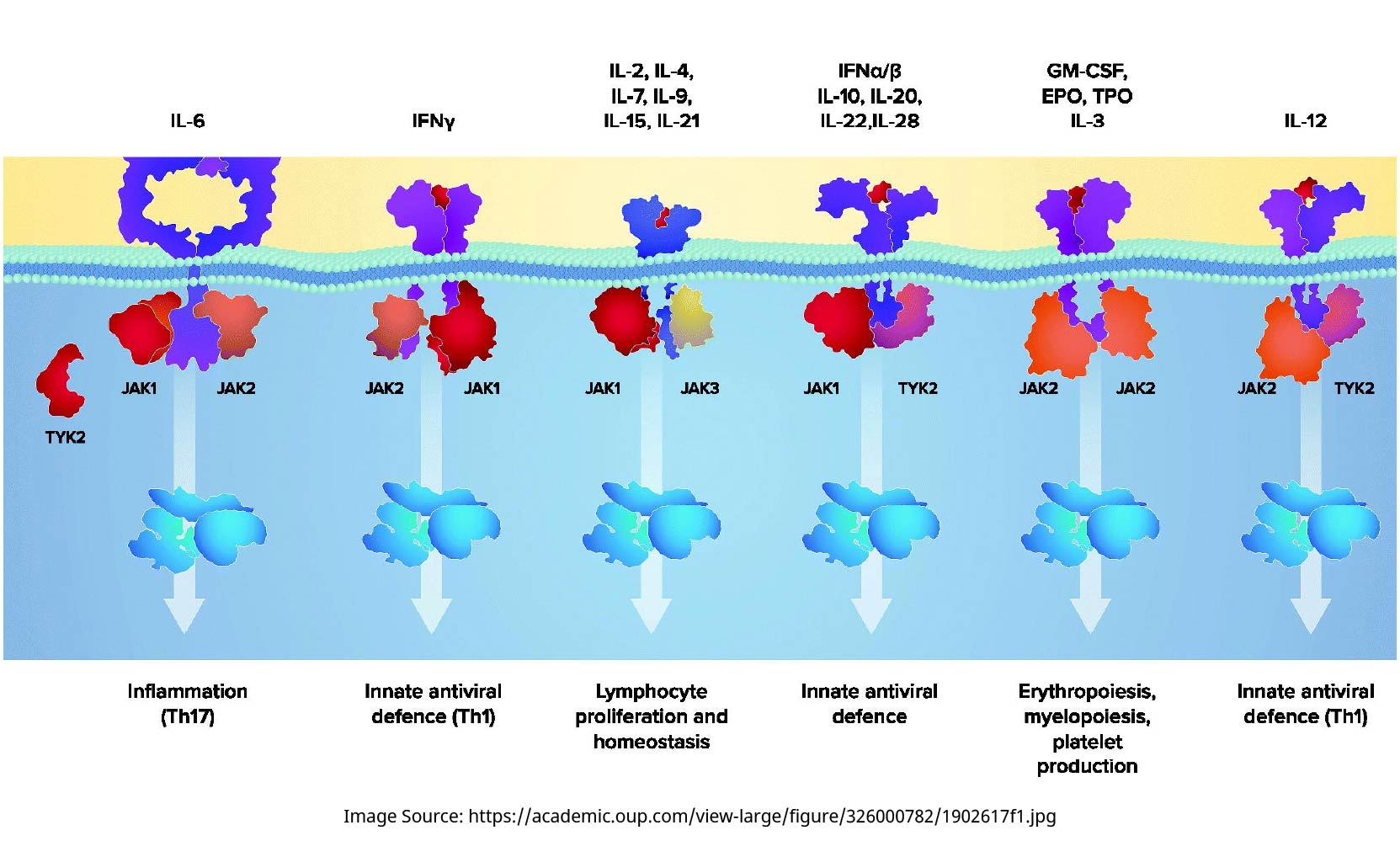
Schematic diagram of the JAK/STAT pathways (adapted from [7]). The four members of the JAK family of enzymes, namely, JAK1, JAK2, JAK3, and TYK2, form homo- and hetero-dimers and one heterotrimer that facilitate the signal transduction of various type I/II cytokines and growth factors implicated in inflammatory and hematopoietic pathways. EPO: erythropoietin; GM-CSF: granulocyte-macrophage colony-stimulating factor; IFN: interferon; IL: interleukin; JAK: Janus kinase; STAT: signal transducer and activator of transcription; Th: T helper cell; TYK: tyrosine kinase.
Jakinibs currently on the market for RA treatment include tofacitinib, baricitinib, upadacitinib, and peficitinib (Japan only). The latest jakinib to enter the international market for RA treatment is filgotinib. A body of clinical evidence has been built up for the efficacy and safety of filgotinib in patients with moderately-to-severely active RA. This article will review the pharmacological properties, efficacy, and safety of filgotinib as demonstrated in clinical studies. Expert opinion will be provided on the various jakinibs for RA treatment from the viewpoints of basic research and clinical practice.
Pharmacology of filgotinib
Pharmacokinetics of filgotinib
In healthy volunteers, the Cmax and AUC0–24 h of filgotinib and its active metabolite increased proportionally with oral dose in the dosing range of 10–200 mg [9]. After a single oral dose of 50–200 mg filgotinib, its mean time to peak plasma concentration and half-life was 2–3 h and 5–6 h, respectively. The active metabolite of filgotinib could be detected in the plasma within 30 min after dosing, peaking in concentration in 3–5 h and exhibiting a half-life of 18–23 h [9]. More than 80% of the elimination of filgotinib and its active metabolite is through the urine [10]. Steady-state PK data from healthy volunteers aged 40–50 years showed that over 24 h, the mean amounts of filgotinib and its active metabolite excreted in the urine were equivalent to about 8% and 34%, respectively, of the daily dose administered [11].
The metabolism of filgotinib is independent of hepatic CYP450, thereby lowering the potential for drug–drug interactions with a number of other agents [10]. For example, filgotinib does not require dose adjustments when used concomitantly with ketoconazole, rifampicin, or probenecid, some of the comedications with notable potential drug-drug interactions to be considered when using jakinibs [12].
Pharmacodynamics of filgotinib
Filgotinib was first identified as a JAK1/JAK2 inhibitor through biochemical assays, while cellular and whole blood assays subsequently showed filgotinib to be approximately 30 fold more selective for JAK1 (IC50: 0.629 µM) than JAK2 (IC50: 17.5 µM) [13]. The main metabolite of filgotinib also exhibited selective activity towards JAK1, with an IC50 of 11.9 µM [9]. STAT phosphorylation assays confirmed that filgotinib inhibited STAT phosphorylation pathways dependent on JAK1-containing JAK heterodimers more strongly than it did those that are JAK2/2 homodimer-dependent [13].
Based on the PK data obtained from phase 1 studies in healthy volunteers, population PK/PD models were constructed for filgotinib and its active metabolite to simulate the inhibition of STAT1 phosphorylation following dosing of filgotinib. For doses below 300 mg, the model structure assumed complete conversion of filgotinib to its active metabolite [9]. The results predicted that maximal PD response would be achieved with a daily dose of 200 mg, which was the highest dose tested in subsequent phase 2b dose-finding studies [9]. Since filgotinib and its metabolite both contribute to the PD effects, the combined, prolonged duration of JAK1 inhibition makes once-daily dosing a feasible dosing frequency, as was later confirmed in phase 2b dose-finding studies.
An integrated model using in vitro cytokine inhibition data and plasma PK data predicted the currently available jakinibs to have similar potency profiles for cytokine receptor inhibition, although the model did show filgotinib and its active metabolite to have lower potency than baricitinib, tofacitinib, and upadacitinib for inhibiting IL-21 (JAK1/JAK3 dependent, implicated in NK cell maintenance) and G-CSF (JAK1/JAK2 dependent, implicated in granulocyte production) signaling [14]. More recent modeling studies went further to examine the time-adjusted activities of jakinibs. Filgotinib together with its active metabolite was calculated to effect 27.3% (at 100 mg) or 45.8% (at 200 mg) mean inhibition of JAK1 over 24 h (by area under the curve [AUC]; Figure 2(A), solid curves) [15]. Using published human PK data and consistent modeling methods, the differential inhibition of JAK1 versus JAK2 was predicted to be higher with filgotinib than with baricitinib, tofacitinib, or upadacitinib (Figure 2(A–D)), each at clinical doses and over a period of 24 h [15]. Based on modeled daily percent STATs inhibition, filgotinib inhibited IFNα/pSTAT5 and IL-6/pSTAT1 to similar extents as the other three jakinibs, consistent with its efficacy in treating RA, while having the least inhibition of IL-2, IL-15, and IL-4 signaling among the four jakinibs, as well as less inhibition of GM-CSF/pSTAT5, IFNγ/pSTAT1, and G-CSF/pSTAT3, especially compared with upadacitinib and baricitinib [16].
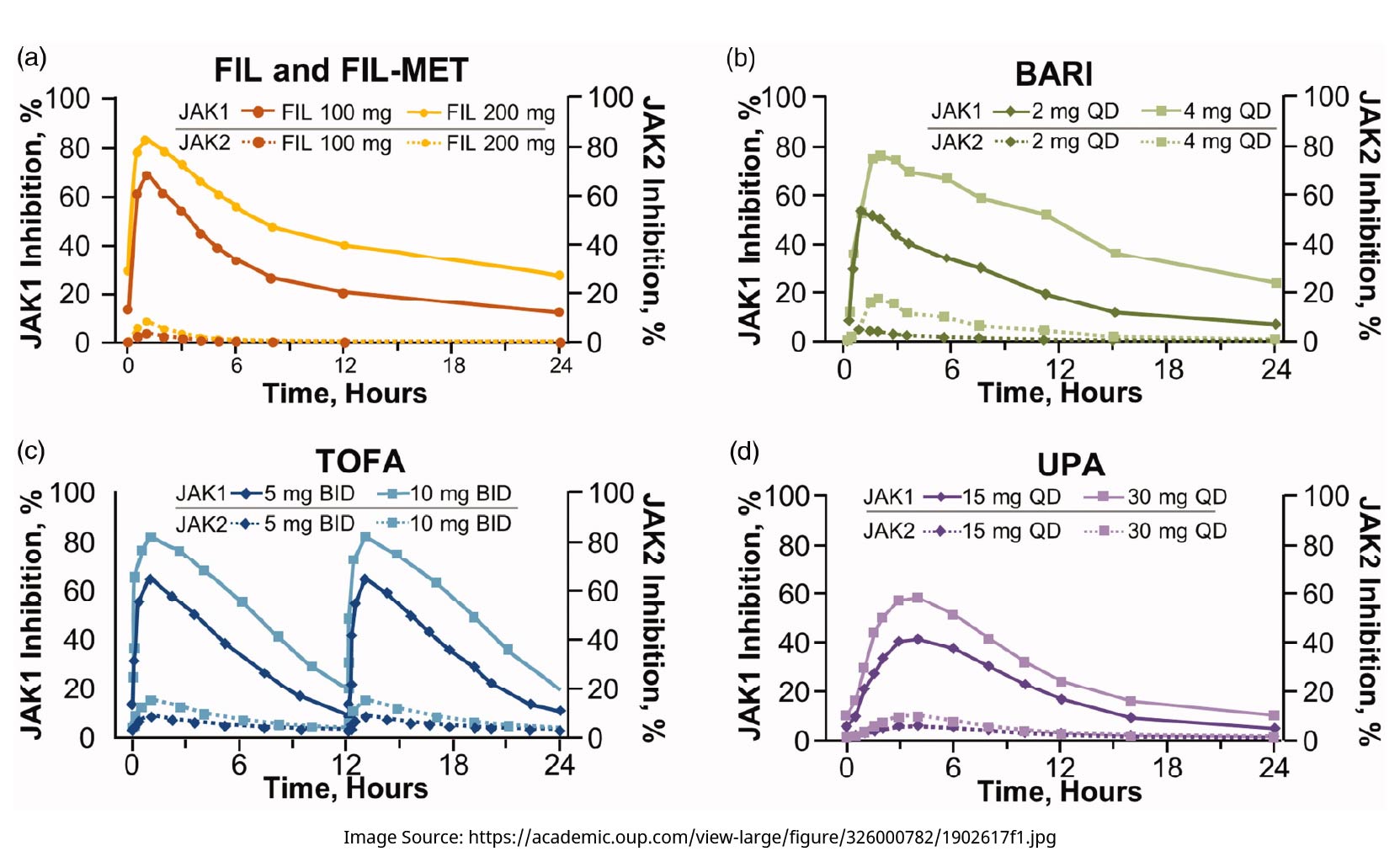
Modeled 24-h pharmacodynamic coverage of JAK1 and JAK2 inhibition by jakinibs at clinical doses [15]. BARI: baricitinib; BID: twice a day; FIL: filgotinib; FIL-MET: active metabolite of filgotinib; JAK: Janus kinase; QD: once a day; TOFA: tofacitinib; UPA: upadacitinib.
Apart from STAT phosphorylation, the potency of filgotinib was assessed in vitro for some other cellular processes [15]: filgotinib inhibited erythroid progenitor expansion less potently (IC50: 1140–1960 nM) compared with tofacitinib (IC50: 110–210 nM) or with baricitinib and upadacitinib (IC50: 25–42 nM). Filgotinib also inhibited IL-15-induced natural killer (NK) cell proliferation less potently (IC50: 315 nM) than the other three jakinibs (IC50: 4–12 nM). Filgotinib inhibited LXR agonist-induced CETP expression (IC50:15.3µM), an inhibitory activity not observed with the other three jakinibs [15]. Using these in vitro potency data and human PK data, PK/PD modeling predicted that at clinical doses, the inhibition of NK cell proliferation by filgotinib would be 13–37% points lower than that by the other jakinibs, and filgotinib would reduce CETP expression by 17–27% [15].
Clinical efficacy
The core clinical program evaluating filgotinib in patients with moderately-to-severely active RA consists of three phase 2b (DARWIN 1–3) and four phase 3 (FINCH 1–4) studies. In accordance with the standard clinical use of targeted therapies, the development scheme of filgotinib mainly focused on its use as combination therapy with methotrexate (MTX) or other csDMARDs in three patient populations defined by treatment history, although filgotinib monotherapy was also evaluated in some studies (Table 1). This section will review the efficacy data from six of these studies, while data are not yet available for the ongoing long-term extension, the FINCH 4 study.
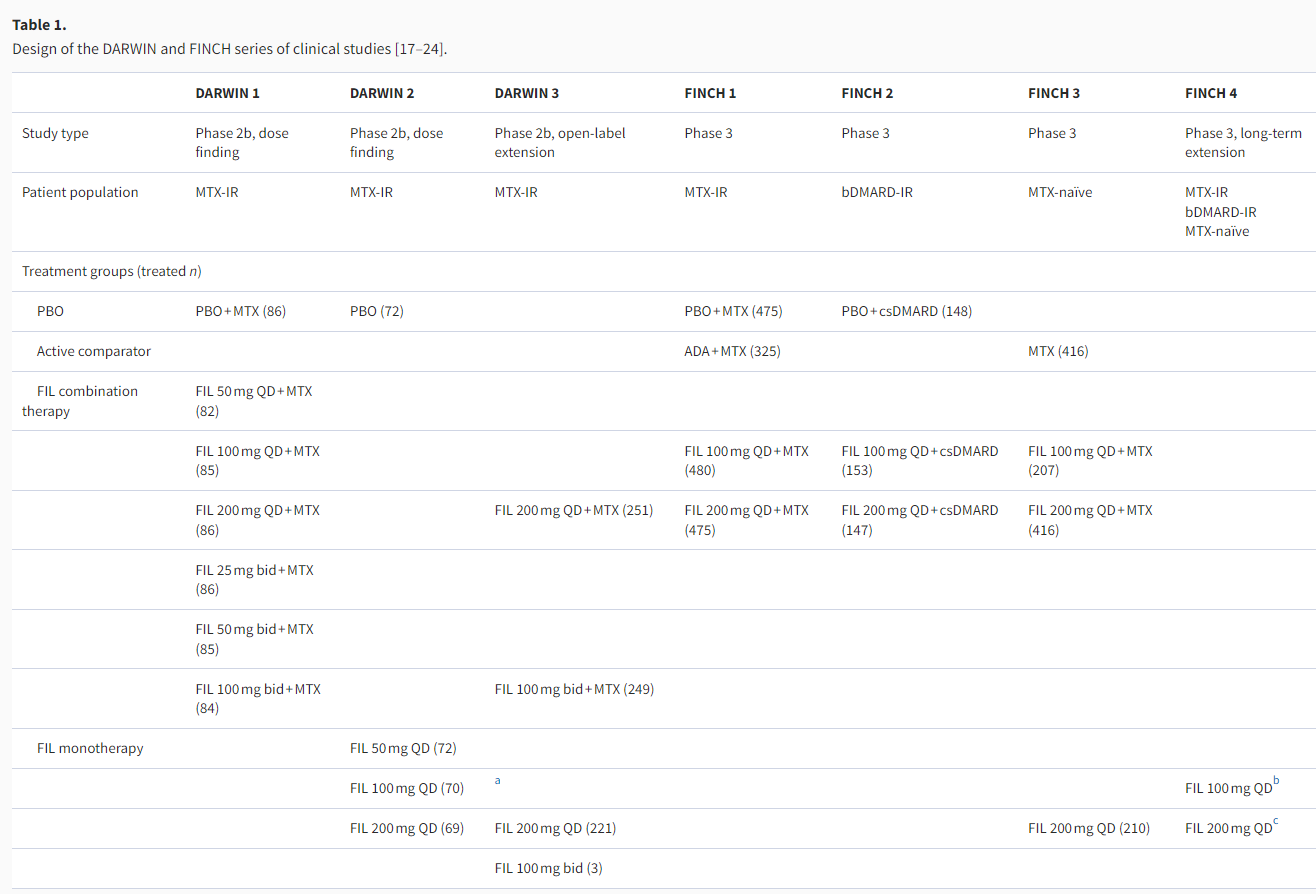
Filgotinib in patients with inadequate responses to methotrexate
The DARWIN 1, DARWIN 3, and FINCH 1 studies evaluated the use of filgotinib in combination with MTX in patients with IRs to MTX as per the second-line therapy recommended in the EULAR treatment algorithm. The key efficacy data are summarized in Table 2.
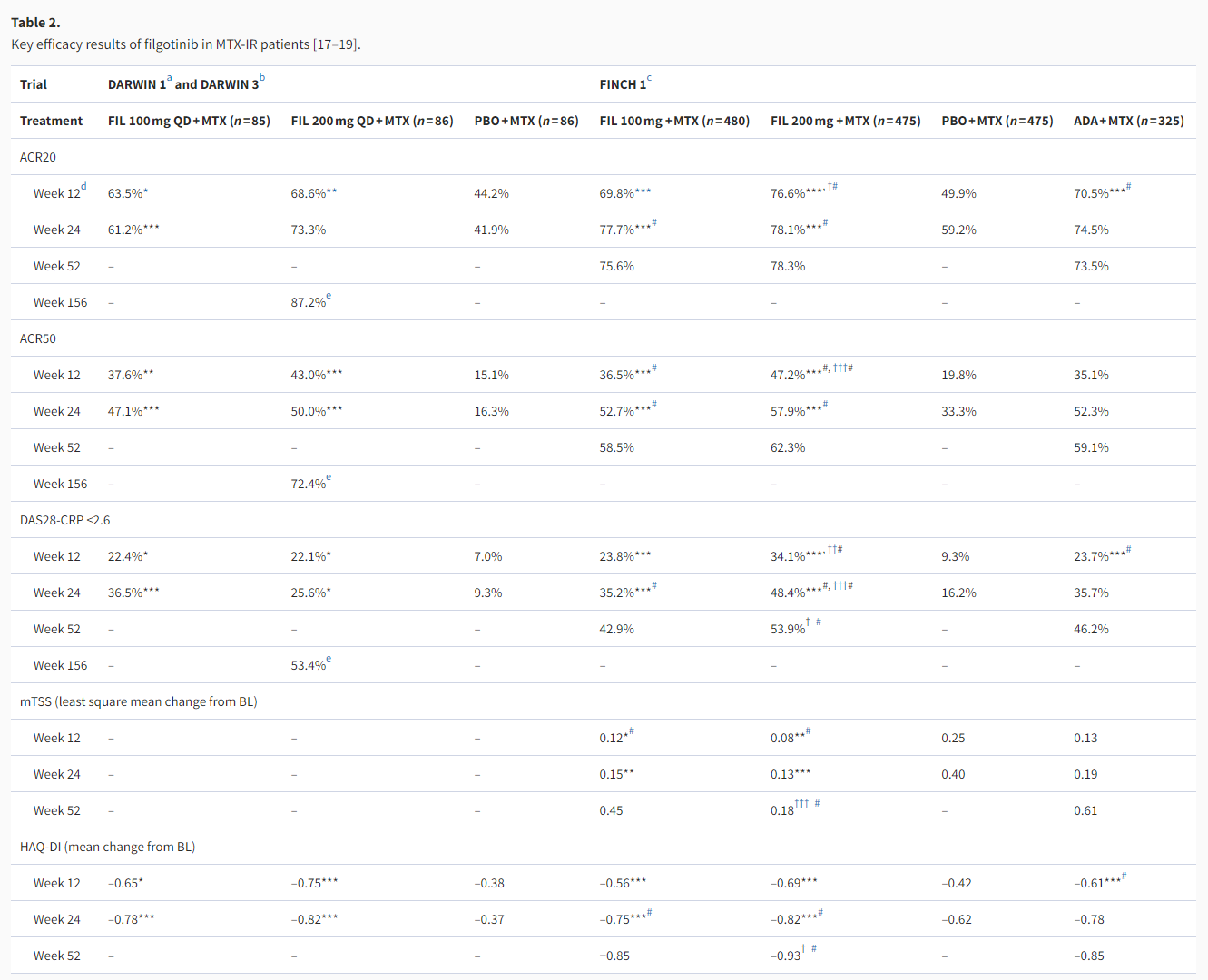
ACR20/50: American College of Rheumatology 20%/50% improvement criteria; ADA: adalimumab; BL: baseline; DAS28-CRP: Disease Activity Score based on 28 joints and C reactive protein value; FAS: full-analysis set; FIL: filgotinib; HAQ-DI: Health Assessment Questionnaire-Disability Index; IR: inadequate response; ITT: intention-to-treat; LOCF: last observation carried forward; MMRM: mixed-effects model for repeated measures; mTSS: modified total Sharp score; MTX: methotrexate; NRI: non-responder imputation; PBO: placebo; QD: once a day.
In the phase 2b, dose-finding study of DARWIN 1, filgotinib 100 and 200 mg achieved efficacy superiority over placebo as measured by the primary endpoint of ACR20 response rate at week 12 (Table 2). For each of the filgotinib daily doses, once- or twice-daily dosing showed no significant difference in efficacy [17]. Filgotinib 100 and 200 mg once-daily have thus been used as the principal dosing in most of the subsequent clinical studies. Among the patients who completed DARWIN 1, 497 entered the DARWIN 3 open-label extension (OLE) study and all received the daily dose of filgotinib 200 mg in combination with MTX. Sustained efficacy was observed through week 156, at which point 87.2% (220/252, observed cases) of patients achieved ACR20 response and 69.0% (138/200, observed cases) of patients had DAS28-CRP ≤3.2 [18]. It should be noted that at week 156, only 59.9% of the 739 patients (from DARWIN 1 and 2) enrolled in DARWIN 3 remained on study treatment, with adverse events (26.5%) and patient requests (9.1%) being the most common reasons for discontinuation [18]. Reflecting this drop-out situation, the week 156 efficacy endpoints were analyzed by observed cases, where the total number of patients with available results among those receiving filgotinib 200 mg plus MTX ranged from n = 133 to 252 for the binary endpoints of ACR20/50/70 response rates and remission/low disease activity (LDA) rates by DAS28-CRP [18].
In the 52-week phase 3 study of FINCH 1, in addition to demonstrating superior efficacy over placebo, filgotinib 100 and 200 mg regimens were compared to the active comparator of adalimumab (40 mg Q2W), a standard-of-care TNFi in RA treatment [19]. At week 12 (primary analysis), higher percentages of patients in the filgotinib 200 mg group achieved ACR20/50/70 responses than in the adalimumab group, with the numerical improvements being 6.1–12.1% points, but statistical significance could not be demonstrated for these differences with only exploratory p values available (Table 2). In terms of disease activity, the filgotinib 200 mg group had higher percentages of patients with DAS28-CRP ≤ 3.2 (49.7% vs. 43.4%, p< .001 for non-inferiority test) and DAS28-CRP <2.6 (34.1% vs. 23.7%, nominal p< .01 for superiority test) compared with the adalimumab group. As for filgotinib 100 mg, the ACR20/50/70 response rates and the percentages of patients with LDA or remission by DAS28-CRP were mostly similar to those with adalimumab (Table 2) [19]. Similar trends of relative efficacy were observed among the three active treatment groups up to week 52 [19]. Change from baseline in van der Heijde modified total Sharp score (mTSS) at week 24 was a key secondary endpoint, which showed both filgotinib 100 and 200 mg as significantly inhibiting radiographic progression compared with placebo, and in exploratory analysis at week 52 filgotinib 200 mg showed less radiographic progression compared with adalimumab (Table 2) [19]. As evident from these results and from Table 2, filgotinib 200 mg tended to achieve numerically better efficacy results than filgotinib 100 mg.
For MTX-IR patients, filgotinib 100 and 200 mg monotherapies have also been shown to be more efficacious than placebo in the phase 2 DARWIN 2 study [20]. DARWIN 2 completers went on to receive filgotinib 200 mg monotherapy in the DARWIN 3 OLE study and showed sustained efficacy. Within DARWIN 3, this group receiving 200 mg filgotinib monotherapy had similar or slightly lower ACR 20/50/70 response rates and DAS28-CRP remission/LDA rates at week 156 compared with the group receiving filgotinib 200 mg + MTX therapy (DARWIN 1 completers) [18].
Filgotinib in patients with inadequate responses to biologic disease-modifying anti-rheumatic drugs
FINCH 2 evaluated the use of filgotinib in combination with csDMARDs in patients with prior bDMARD failure or intolerance, i.e. as per the third-line therapy recommended in the EULAR treatment algorithm. In this 24-week phase 3 study, patients with IRs or intolerance to ≥1 prior bDMARD were randomized to receive placebo, filgotinib 200 mg, or filgotinib 100 mg. Results of primary and key secondary endpoints supported the superior efficacy of both filgotinib doses vs. placebo (Table 3) [21]. Subgroup analyses showed that the efficacy of filgotinib was not affected by the number or mechanism of action (MOA) of prior bDMARDs, as patients with ≥3 prior bDMARDs or ≥1 MOA of prior bDMARDs, as well as those previously exposed to IL-6 inhibitors or TNFis, all achieved efficacy outcomes comparable to the overall study population [21,25,26]. Again, it was noted that the filgotinib 200 mg group tended to have numerically better results than the 100 mg group for many, though not all, efficacy endpoints. The study did not include radiographic endpoints to evaluate structural joint damage.
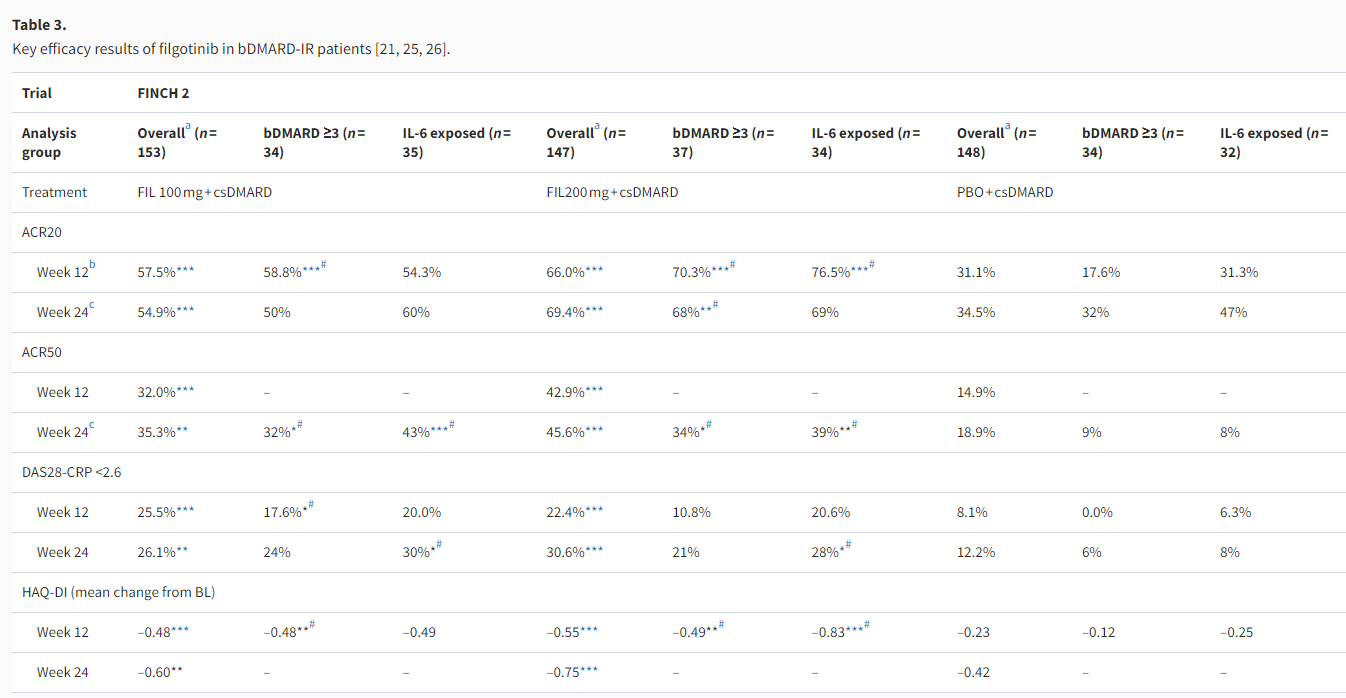
ACR20/50: American College of Rheumatology 20%/50% improvement criteria; (b/cs)DMARD: (biologic/conventional synthetic) disease-modifying anti-rheumatic drug; BL: baseline; DAS28-CRP: Disease Activity Score based on 28 joints and C reactive protein value; FAS: full-analysis set; FIL: filgotinib; HAQ-DI: Health Assessment Questionnaire-Disability Index; IL-6: interleukin-6; IR: inadequate response; MMRM: mixed-effects model for repeated measures; mTSS: modified total Sharp score; MTX: methotrexate; NRI: non-responder imputation; PBO: placebo.
Filgotinib in methotrexate-naïve patients
FINCH 3 evaluated the use of filgotinib in MTX-naïve patients. Primary (24-week) data showed that filgotinib 100 or 200 mg in combination with MTX achieved significantly higher ACR20 than MTX monotherapy (Table 4) [22]. With filgotinib 200 mg monotherapy, higher proportions of patients achieved ACR50/70 at week 24 than with MTX monotherapy, although the ACR20 response rate was numerically but not statistically significantly higher [22]. For both mono- and combination therapies, filgotinib-treated groups showed efficacy up to week 52. In terms of structural damage progression, all filgotinib-treated groups had smaller increases in mTSS than the MTX monotherapy group at weeks 24 and 52, although only nominal p values were available (Table 4). At week 24, filgotinib-treated groups had 77–83% of patients classified as having no radiographic progression (change in mTSS ≤0), compared with 73% in the MTX monotherapy group [22]. Among the three filgotinib-treated arms, filgotinib 200 mg combination therapy appeared to give the highest numerical results for most efficacy endpoints.
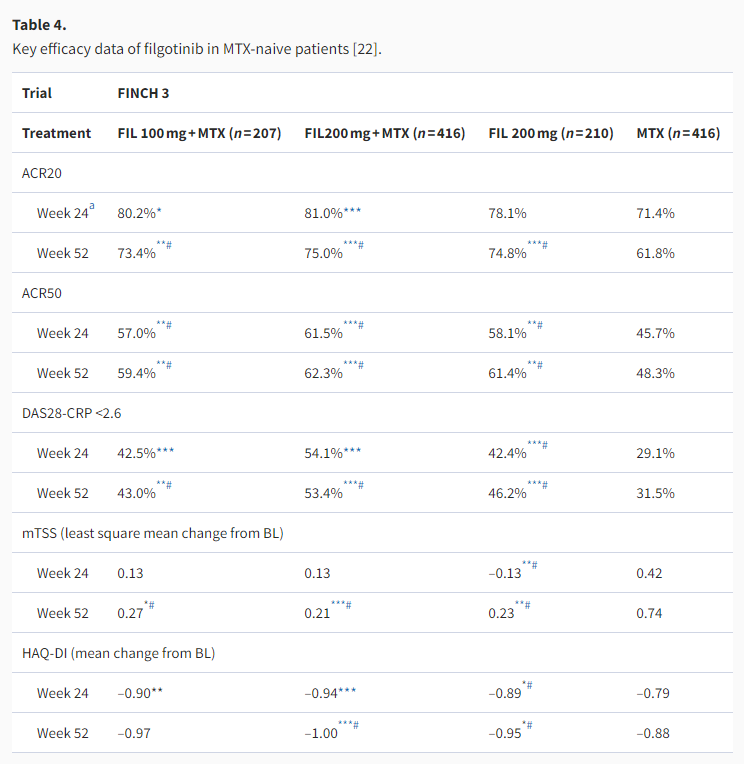
Analysis based on all randomized patients who received ≥1 dose of study drug with NRI for ACR20/50 and DAS28-CRP <2.6, analysis by a linear mixed-effects model for mTSS, and analysis by MMRM for HAQ-DI.
ACR20/50: American College of Rheumatology 20%/50% improvement criteria; BL: baseline; DAS28-CRP: Disease Activity Score based on 28 joints and C reactive protein value; FAS: full-analysis set; FIL: filgotinib; HAQ-DI: Health Assessment Questionnaire-Disability Index; IL-6: interleukin-6; IR: inadequate response; MMRM: mixed-effects model for repeated measures; mTSS: modified total Sharp score; MTX: methotrexate; NRI: non-responder imputation; PBO: placebo.
Safety and tolerability of filgotinib
In the primary and full-duration results of the individual clinical studies, filgotinib appeared to consistently exhibit acceptable safety and tolerability [17–22]. An integrated safety analysis of the seven phase 2b/3 clinical studies of filgotinib in RA is underway, which to date has accrued a total of 4544.5 patient-years (PY) of filgotinib exposure [27]. Currently available results showed that during the corresponding controlled study periods (12 weeks for placebo, 52 weeks for MTX, and 52 weeks for adalimumab), the incidence of TEAEs of the filgotinib 100 and 200 mg groups was generally comparable with that of the placebo- or active comparator-treated groups, with low incidences of SAEs and AEs leading to discontinuation [27]. Additionally, the as-treated incidence of safety events was calculated for filgotinib-treated patients from all the individual studies to assess if any differences could be detected between the two doses and with longer exposures. As summarized in Table 5, the safety results from 3079.2 PY of exposure to filgotinib 200 mg were comparable to those from 1465.3 PY of exposure to filgotinib 100 mg, revealing no clear dose-dependent elevation of safety risks [27]. Longer-term data from clinical and real-world settings would be needed to further verify the comparative safety profiles of the two doses of filgotinib.
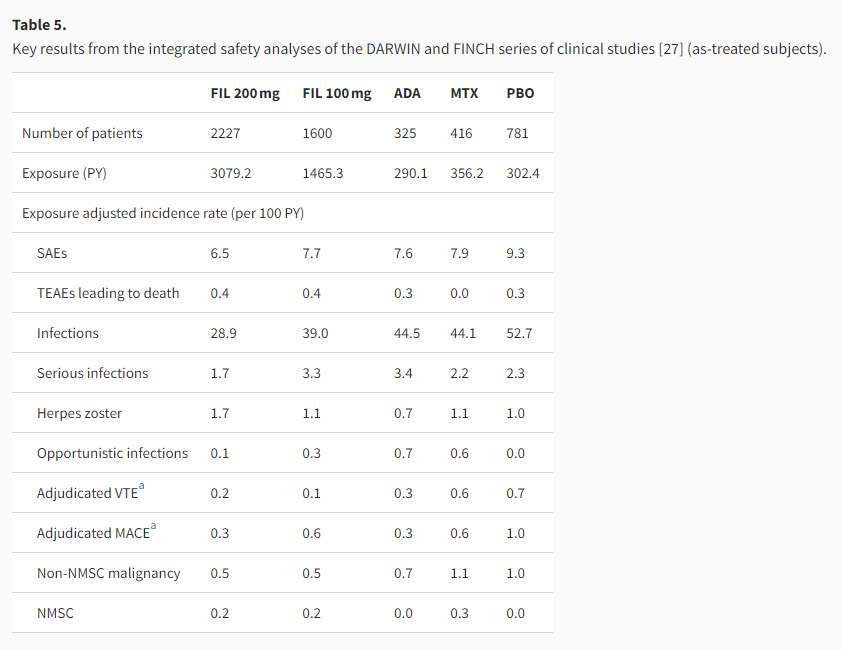
ADA: adalimumab; FIL: filgotinib; MACE: major adverse cardiovascular events; MTX: methotrexate; NMSC: non-melanoma skin cancer; PBO: placebo; PY: patient-year; SAE: serious adverse event; TEAE: treatment-emergent adverse event; VTE: venous thromboembolism.
Regarding opportunistic infections, the 52-week incidence was 0/100 PY for filgotinib 100 and 200 mg regimens, and the as-treated incidence was 0.3/100 and 0.1/100 PY for the two filgotinib doses respectively, all of which were lower than the 52-week incidence for adalimumab (0.7/100 PY) or MTX (0.5/100 PY) [27]. The as-treated incidence of herpes zoster infections was 1.1/100 and 1.7/100 PY for filgotinib 100 and 200 mg regimens, respectively, while the 52-week incidence for adalimumab and MTX was 0.7/100 and 1.1/100 PY, respectively [27]. Asian ethnicity has been previously correlated with a higher risk for herpes zoster infections among RA patients, but the pooled safety analysis of Japanese patients enrolled in FINCH 1–3 reported that the percentage of filgotinib-treated patients experiencing herpes zoster infection was low at 0.6%, similar to those for adalimumab- and placebo-treated patients (0.6% and 0.4%, respectively) [28]. For bDMARD-IR patients, the subgroup analysis of Japanese patients from FINCH 2 reported one case of herpes zoster infection in a patient treated with filgotinib 200 mg among a total of 27 patients receiving filgotinib over 24 weeks [29].
The as-treated incidence of centrally adjudicated major adverse cardiovascular events (MACE) was 0.6/100 and 0.3/100 PY for filgotinib 100 and 200 mg, respectively, similar to the 52-week incidence for adalimumab (0.3/100 PY) and MTX (0.5/100 PY). As for centrally adjudicated venous thromboembolism (VTE) events, the as-treated incidence for filgotinib 100 mg (0.1/100 PY) and 200 mg (0.2/100 PY) was slightly lower than that for 52-week adalimumab (0.3/100 PY) or MTX (0.5/100 PY) [27].
Filgotinib-treated patients exhibited consistent patterns of hematological changes, including dose-dependent increase in hemoglobin, clinically insignificant decrease in neutrophil counts, small decline in platelet counts that typically stabilized after 4 weeks of treatment, and no on-treatment reduction in lymphocyte or NK cell counts. Filgotinib-treated patients exhibited greater increases in HDL versus LDL, resulting in decreased (or stable) LDL:HDL ratio [17,19–22].
Patients treated with filgotinib experienced transient elevation in serum creatinine, as well as mild liver enzyme elevation, without signs of drug-induced hepatocellular injury. Overall, filgotinib exhibited an acceptable adverse effect profile on the hepatic and renal function of patients with RA [17,19–22].
The FINCH 1–3 studies reported that serum creatine phosphokinase (CPK) elevation occurred more frequently in filgotinib-treated patients than in placebo-treated patients; Grade ≥3 CPK elevation events were few, reported by ≤2% of patients in any filgotinib treatment arm [19,21,22].
As stated in the Japanese and European drug labels of filgotinib dated September 2020, impaired spermatogenesis and histopathological changes on the testes and epididymis were observed in pre-clinical animal studies, albeit at exposure levels 5.1 times (in dogs) and 7.3 times (in rats) that of 200 mg once a day (QD) in humans [30,31]. It is unknown if this could occur in humans and in August 2020 the US FDA requested data from two ongoing studies to evaluate this question before completing its review of the NDA [32]: the MANTA (NCT03201445) and MANTA-Ray (NCT03926195) studies were designed to assess whether filgotinib has an impact on sperm parameters for adult males with IMIDs. Such preclinical findings or regulatory requests have not been observed for other jakinibs and, therefore, it is unlikely that this observation is a class-related effect. Additionally, in the absence of in vivo data, it is difficult to draw conclusions on the potential for filgotinib to impact spermatogenesis or fertility in these patients. However, until such evidence becomes available the potential risk should be discussed with male patients [30].
Expert opinion on jakinibs (from US, EU, and Japan): a basic research perspective
The pathogenesis of RA is not yet fully understood, and the existing picture is one of complex signaling crosstalk with network effects and plethora of cytokines implicated [33]. As such, treatment efficacy may arise from broad-spectrum cytokine inhibition that simultaneously suppresses multiple players in the pathogenic signaling network. Oral agents like jakinibs that target signaling molecules in nodal positions integrating diverse upstream cytokines and downstream effectors thus represent a promising therapeutic tool.
Theoretically, the ‘ideal’ JAK inhibition should aim to block only pathogenic signaling for RA without impacting the other JAK-dependent, physiological signaling processes, if such an outcome is possible. Thus far we have not identified an RA-specific JAK dependency and as such the more selective approaches are focused mainly on perceived longer-term safety benefits and the chance thereby to increase the degree of inhibition achievable. In the consequential search for more selective jakinibs, in vitro assessments demonstrated that the ability to modulate distinct cytokine pathways appears to differ among the currently available jakinibs. However, in their respective pre-clinical studies, the in vitro assays used to obtain selectivity data were not necessarily consistent across different jakinibs [13,34,35]. Side-by-side comparisons of jakinibs in cell-based/whole blood assays have shown that for a given jakinib, its pre-clinical fold-selectivity data for individual JAK isoforms could not be used to easily deduce its ability to inhibit specific cytokine signaling pathways [16,36]. For example, an earlier study comparing tofacitinib, baricitinib, and upadacitinib reported that all three inhibited JAK2/2-dependent cytokine signaling pathways, with upadacitinib and baricitinib showing higher potency than tofacitinib [36], while a more recent study reported baricitinib as showing lower JAK1 selectivity (≤5.1 fold versus non-JAK1 pathways), whereas tofacitinib, upadacitinib, and filgotinib showed >5-fold selectivity for JAK1-dependent pathways over JAK2-dependent pathways [16].
The results of in vitro assays are highly dependent on factors such as the doses of JAK inhibitors, the cytokine stimulations used, the STATs chosen for phosphorylation assessment, the cell types used in cell-based assays, and others. Jakinibs also cannot be expected to potently and/or continuously inhibit an individual cytokine pathway for certain periods (e.g. 24 h) according to the PD indices obtained in in vitro assays [36], which do not recapitulate the complexity of in vivo immune modulation by jakinibs. In addition to the intricacy captured in Figure 1, JAK-dependent cytokine signaling in vivo is further influenced by the individual genetics (e.g. single nucleotide polymorphisms of STAT isoforms), the tissue penetration and/or distribution of drugs, the intrinsic JAK expression patterns in targeted cell types and redundant intercellular communications at the site of inflammation, the cytokine and inflammation environment (e.g. that during activation phase versus fibrosis phase), as well as the dynamic balance between Th17 and Treg cells.
With the multiple layers of complexity discussed above, it proves challenging to make direct, mechanistic associations between the selectivity of jakinibs and their clinical efficacy and safety profiles. In the case of filgotinib, some hypothesized that its ability to maintain preferential JAK1 inhibition over JAK2 at high doses may underly its dose-dependent incremental efficacy in the absence of dose-limiting side effects; nevertheless, further study would be needed to provide definitive mechanistic support for this notion. Data from DARWIN 1–2 showed that filgotinib regulated biomarkers for RA-related immune response, matrix degradation, angiogenesis, and leukocyte adhesion and recruitment, in correlation with reduction in disease activity [37]. More such studies of cytokine and immune cell profiling may help shed light on the association between jakinib selectivity, specific cytokine signaling pathways in RA pathogenesis, and clinical outcomes of RA treatment with jakinibs.
Expert opinion on jakinibs (from US, EU, and Japan): a clinical perspective
As more treatment options become available, the question of how different jakinibs might compare with one another becomes increasingly pertinent for guiding clinical decisions. In an attempt to address this, some recent publications tabulated, side-by-side, the efficacy and safety data of different jakinibs from their respective phase 2/3 trials that were not done head-to-head [2,8,38]. Of course, such comparisons are tenuous at best. Some have tried to use network meta-analyses to try to allow indirect comparison, but such assessments have their own limitations [39–43]. Only properly powered head-to-head clinical trials can determine whether there are clinically meaningful differences in efficacy and safety among multiple jakinibs. This is also applicable when considering the effects of jakinibs on patient-reported outcomes (PROs) in patients with RA, as presently PRO data are only available from phase 2/3 studies of individual jakinibs while between-regimen comparison is lacking. However, the sample size and follow-up duration required to power the detection of small differences may make head-to-head clinical trials practically challenging to conduct. Clinicians may thus value real-world evidence more for validating the long-term safety and tolerability of the newer jakinibs. As seen with DARWIN 3, long-term follow-up is prone to higher drop-out rates even in clinical trials, while in real-world settings many patients may taper or discontinue targeted therapies quite soon due to financial and reimbursement constraints. These factors would need to be considered when designing future clinical and real-world studies for filgotinib and other jakinibs.
With bDMARDs, a considerable proportion of patients discontinue treatment due to intolerance or loss of response. Jakinibs offer some advantages over other agents, but RA patients treated with jakinibs may still be prone to certain complications or adverse events. JAK inhibition may decrease hemoglobin and increase the risk of anemia. The risk of infections, particularly that of herpes zoster infection, can be elevated due to the impact of JAK inhibition on the immune system. Potentially serious events such as malignancy and thromboembolic events have also been reported with jakinibs. Side-by-side tabulation of across-trial data, which it is important to note were not head-to-head comparisons have suggested that filgotinib might have lower incidence rates of herpes zoster infection and venous thrombotic events than other jakinibs approved to date [8,38]. Nevertheless, as mentioned above, whether filgotinib has a more favorable safety profile remains to be demonstrated definitively and at this stage should be considered with caution in clinical practice.
Besides effectiveness and safety, drug cost is also an important factor influencing the choice of therapies in clinical practice. The predominant pattern of using targeted therapies is still as second- or third-line treatment in combination with csDMARDs, as dictated by treatment guidelines and reimbursement requirements. However, many RA patients may require access to different combinational or sequential regimens and long-term treatment to successfully control the disease [5]. Whether the use of jakinibs can be expanded beyond the established pattern and form part of more versatile treatment regimens suitable for personalized medicine would await further research.
In clinical studies, filgotinib 200 mg tended to give better efficacy results than filgotinib 100 mg, while the two doses generally showed comparable safety and tolerability profiles. Logically consistent with these observations, the Japanese and European approval of filgotinib recommended 200 mg QD as the standard dose. In Europe, filgotinib 100 mg QD is reserved for patients with creatinine clearance of 15 to <60 mL/min, and for patients aged ≥75 years as a starting dose. Similarly in Japan, filgotinib 100 mg QD is recommended for patients with eGFR of 15–60 mL/min/1.73 m2, or when otherwise deemed suitable depending on individual patient’s condition. Conceivably, filgotinib 100 mg may also be a convenient tool for the procedure of drug tapering when deemed appropriate for patients in persistent remission.
It is also worth noting that some jakinibs have demonstrated efficacy towards other IMIDs than RA. Upadacitinib showed clinical efficacy in phase 2/3 trials for atopic dermatitis [44], ulcerative colitis [44], Crohn’s disease [45], psoriatic arthritis [46], and ankylosing spondylitis [47]. Likewise, positive phase 2/3 results have been reported for filgotinib in treating Crohn’s disease [44], ulcerative colitis [48], psoriatic arthritis [49], and ankylosing spondylitis [50]. Tofacitinib has received regulatory approval for treating psoriatic arthritis and ulcerative colitis [51], and has positive data in ankylosing spondylitis and also a number of dermatologic conditions. These features not only make jakinibs more useful clinically for treating broader patient populations and patients suffering from multiple, comorbid IMIDs, but also point to potentially interesting research questions about the roles of JAK signaling in the pathogenesis of a wide variety of IMIDs.
Conclusion
In in vitro assays, filgotinib and its active metabolite preferentially inhibited JAK1-dependent cytokine signaling pathways rather than JAK2/2 homodimer-dependent pathways. The efficacy of filgotinib in combination with csDMARDs has been demonstrated in patients with moderately-to-severely active RA with IRs to MTX (DARWIN 1 and 3; FINCH 1) or prior bDMARD treatments (FINCH 2), supporting its use as later-line treatments in accordance with international RA management guidelines. The use of filgotinib as monotherapy and as first-line treatment in MTX-naïve patients have also been evaluated with positive outcomes (FINCH 3). In clinical studies, filgotinib consistently showed acceptable safety and tolerability profiles, including those concerning known adverse events associated with jakinibs such as opportunistic infections, MACE and VTE, and hematological changes; meanwhile, evidence is being sought to clarify the potential effect of filgotinib on spermatogenesis and male fertility in humans. Thus far in clinical studies, filgotinib 200 mg QD tended to show higher efficacy than filgotinib 100 mg QD, with apparently similar profiles of safety and tolerability. As more jakinibs become available for treating RA, more evidence would be needed to elucidate if there are clinically significant differences among them in terms of efficacy and safety. Research into the possible mechanistic association between the JAK isoform/pathway selectivity of jakinibs and their clinical efficacy and safety profiles has also gathered interest, though much remains nebulous due to our partial understanding of RA pathogenesis at present.
Acknowledgments
The authors acknowledge Bo Lyu, PhD, from Costello Medical, Singapore (funded by Gilead Sciences), for medical writing and editorial assistance based on the authors’ input and direction.
Author contributions
Substantial contributions to review conception: Yoshiya Tanaka, Arthur Kavanaugh, Jason Wicklund, and Iain B. McInnes; substantial contributions to interpretation of reviewed literature: Yoshiya Tanaka, Arthur Kavanaugh, Jason Wicklund, and Iain B. McInnes; drafting the article or revising it critically for important intellectual content: Yoshiya Tanaka, Arthur Kavanaugh, Jason Wicklund, and Iain B. McInnes; final approval of the version of the article to be published: Yoshiya Tanaka, Arthur Kavanaugh, Jason Wicklund, and Iain B. McInnes.
Conflicts of interest
Y. Tanaka has received speaking fees and/or honoraria from Daiichi-Sankyo, Eli Lilly, Novartis, YL Biologics, Bristol-Myers, Eisai, Chugai, AbbVie, Astellas, Pfizer, Sanofi, Asahi-kasei, GSK, Mitsubishi-Tanabe, Gilead, Janssen and has received research grants from AbbVie, Mitsubishi-Tanabe, Chugai, Asahi-Kasei, Eisai, Takeda, Daiichi-Sankyo.
A. Kavanaugh has received honoraria and/or conduction clinical research sponsored by Abbvie, Amgen BMS, Eli Lilly, Gilead, Janssen, Novartis, Pfizer.
J. Wicklund is an employee of Gilead Sciences.
I.B. McInnes has received speaking fees and/or honoraria from Abbvie, BMS, Lilly, Gilead, Janssen, Novartis, Celgene, Cabaletta, Astra Zeneca, GSK, Compugen
References:
1. Deane KD, Holers VM. The natural history of rheumatoid arthritis. Clin Ther. 2019;41(7):1256–69.
2. Angelini J, Talotta R, Roncato R, Fornasier G, Barbiero G, Dal Cin L, et al. JAK-inhibitors for the treatment of rheumatoid arthritis: a focus on the present and an outlook on the future. Biomolecules 2020;10(7):1002.
3. O’Shea JJ, Kontzias A, Yamaoka K, Tanaka Y, Laurence A. Janus kinase inhibitors in autoimmune diseases. Ann Rheum Dis. 2013;72 (2):ii111–5.
4. Tanaka Y, Maeshima K, Yamaoka K. In vitro and in vivo analysis of a JAK inhibitor in rheumatoid arthritis. Ann Rheum Dis. 2012;71(2):i70–4.
5. Smolen JS, Landewe RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. 2020;79(6):685–99.
6. Schwartz DM, Bonelli M, Gadina M, O’Shea JJ. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol. 2016;12(1):25–36.
7. Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol. 2017;13(4):234–43.
8. Sunzini F, McInnes I, Siebert S. JAK inhibitors and infections risk: focus on herpes zoster. Ther Adv Musculoskelet Dis. 2020;12:1759720X20936059.
9. Namour F, Diderichsen PM, Cox E, Vayssiere B, Van der Aa A, Tasset C, et al. Pharmacokinetics and pharmacokinetic/pharmacodynamic modeling of filgotinib (GLPG0634), a selective JAK1 inhibitor, in support of phase IIB dose selection. Clin Pharmacokinet. 2015;54(8):859–74.
10. Namour F, Desrivot J, Van der Aa A, Harrison P, Tasset C, van’t Klooster G. Clinical confirmation that the selective JAK1 inhibitor filgotinib (GLPG0634) has a low liability for drug-drug interactions. Drug Metab Lett. 2016;10(1):38–48.
11. Namour F, Fagard L, Van der Aa A, Harrison P, Xin Y, Tasset C. Influence of age and renal impairment on the steady state pharmacokinetics of filgotinib, a selective JAK1 inhibitor. Br J Clin Pharmacol. 2018;84(12):2779–89.
12. Nash P, Kerschbaumer A, Dorner T, Dougados M, Fleischmann RM, Geissler K, et al. Points to consider for the treatment of immune-mediated inflammatory diseases with Janus kinase inhibitors: a consensus statement. Ann Rheum Dis. 2021;80(1):71–87.
13. Van Rompaey L, Galien R, van der Aar EM, Clement-Lacroix P, Nelles L, Smets B, et al. Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J Immunol. 2013;191(7):3568–77.
14. Dowty ME, Lin TH, Jesson MI, Hegen M, Martin DA, Katkade V, et al. Janus kinase inhibitors for the treatment of rheumatoid arthritis demonstrate similar profiles of in vitro cytokine receptor inhibition. Pharmacol Res Perspect. 2019;7(6):e00537.
15. Han P, Meng A, Mollova N, Yu Y, Di Paolo JA. In vitro mechanistic studies demonstrate filgotinib activity that has potential implications for differentiation among JAK inhibitors, EULAR; Madrid, Spain. Ann Rheum Dis 2019;78(S2):A276.
16. Traves PG, Murray B, Campigotto F, Galien R, Meng A, Di Paolo JA. JAK selectivity and the implications for clinical inhibition of pharmacodynamic cytokine signaling by filgotinib, upadacitinib, tofacitinib, and baricitinib. Ann Rheum Dis 2021 DOI:10.1136/annrheumdis-2020-219012.
17. Westhovens R, Taylor PC, Alten R, Pavlova D, Enriquez-Sosa F, Mazur M, et al. Filgotinib (GLPG0634/GS-6034), an oral JAK1 selective inhibitor, is effective in combination with methotrexate (MTX) in patients with active rheumatoid arthritis and insufficient response to MTX: results from a randomised, dose-finding study (DARWIN 1). Ann Rheum Dis. 2017;76(6):998–1008.
18. Kavanaugh A, Westhovens R, Winthrop K, Lee S, Greer J, DeZure A, et al. Rheumatoid arthritis treatment with filgotinib: week 156 safety and efficacy data from a phase 2b open-label extension study. ACR/ARP Annual Meeting. Atlanta, Georgia, 2019.
19. Combe B, Kivitz A, Tanaka Y, van der Heijde D, Simon JA, Baraf HSB, et al. Filgotinib versus placebo or adalimumab in patients with rheumatoid arthritis and inadequate response to methotrexate: a phase III randomised clinical trial. Ann Rheum Dis 2021.
20. Kavanaugh A, Kremer J, Ponce L, Cseuz R, Reshetko OV, Stanislavchuk M, et al. Filgotinib (GLPG0634/GS-6034), an oral selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: results from a randomised, dose-finding study (DARWIN 2). Ann Rheum Dis. 2017;76(6):1009–19.
21. Genovese MC, Kalunian K, Gottenberg JE, Mozaffarian N, Bartok B, Matzkies F, et al. Effect of filgotinib vs. placebo on clinical response in patients with moderate to severe rheumatoid arthritis refractory to disease-modifying antirheumatic drug therapy: the FINCH 2 randomized clinical trial. JAMA 2019;322(4):315–25.
22. Westhovens R, Rigby WFC, van der Heijde D, Ching DWT, Stohl W, Kay J, et al. Filgotinib in combination with methotrexate or as monotherapy versus methotrexate monotherapy in patients with active rheumatoid arthritis and limited or no prior exposure to methotrexate: the phase 3, randomised controlled FINCH 3 trial. Ann Rheum Dis 2021. DOI:10.1136/annrheumdis-2020-219213.
23. Kavanaugh A, Genovese M, Winthrop K, Greenwald M, Ponce L, Sosa F, et al. Rheumatoid arthritis treatment with filgotinib: Week 132 safety data from a phase 2b open-label extension study, ACR/ARP Annual Meeting, Chicago, IL, 2018 (Poster 2551).
24. ClinicalTrials.gov. NCT03025308, Long term extension study to assess the safety and efficacy of filgotinib in adults with rheumatoid arthritis (FINCH 4). [2021-01-27] Available from: https://clinicaltrials.gov/ct2/show/NCT03025308.
25. Genovese M, de Vlam K, Gottenberg JE, Bartok B, Tiamiyu I, Guo Y, et al. A subgroup analysis of clinical efficacy response and quality of life outcomes from phase 3 study of filgotinib in patients with inadequate response to biologic dMARDs. ACR/ARP Annual Meeting, Atlanta, Georgia, 2019.
26. Gottenberg JE, Buch MH, Caporali R, Wright GC, Takeuchi T, Kalunian K, et al. A subgroup analysis of low disease activity and remission from phase 3 study of filgotinib in patients with inadequate response to biologic DMARDs. EULAR, E-Congress, 2020 (Poster THU0204).
27. Genovese M, Winthrop K, Tanaka Y, Takeuchi T, Kivitz A, Matzkies F, et al. Integrated safety analysis of filgotinib treatment for rheumatoid arthritis from 7 clinical trials. EULAR, E-Congress, 2020 (Poster THU0202).
28. Tanaka Y, Keystone EC, Winthrop K, Genovese M, Combe B, Kivitz A, et al. Pooled safety analyses from phase 3 studies of filgotinib in patients with rheumatoid arthritis. Mod Rheumatol 2020;30:S180.
29. Takeuchi T, Matsubara T, Atsumi T, Amano K, Ishiguro N, Sugiyama E, et al. Efficacy and safety of filgotinib in Japanese patients with refractory rheumatoid arthritis: subgroup analyses of a global phase 3 study (FINCH 2). Mod Rheumatol 2021;31:1–16. DOI:10.1080/14397595.2020.1859675.
30. Gilead Sciences. Jyseleca (filgotinib) summary of product information (published 28/09/2020). [2020-11-09] Available from:https://www.ema.europa.eu/en/documents/product-information/jyseleca-epar-product-information_en.pdf.
31. Gilead Sciences. Jyseleca (filgotinib) medical product interview form (based on package insert revised in November 2020) (in Japanese). [2021-03-02] Available from: https://www.jyseleca.jp/-/media/Files/Filgotinib/product/basic/jys_if.pdf.
32. Gilead receives complete response letter for filgotinib for the treatment of moderately to severely active rheumatoid arthritis . 2020. [2020-09-22] Available from: https://www.businesswire.com/news/home/20200818005811/en/.
33. McInnes IB, Buckley CD, Isaacs JD. Cytokines in rheumatoid arthritis – shaping the immunological landscape. Nat Rev Rheumatol. 2016;12(1):63–8.
34. Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. 2010;184(9):5298–307.
35. Parmentier JM, Voss J, Graff C, Schwartz A, Argiriadi M, Friedman M, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. 2018;2:23.
36. McInnes IB, Byers NL, Higgs RE, Lee J, Macias WL, Na S, et al. Comparison of baricitinib, upadacitinib, and tofacitinib mediated regulation of cytokine signaling in human leukocyte subpopulations. Arthritis Res Ther. 2019;21(1):183.
37. Tarrant JM, Galien R, Li W, Goyal L, Pan Y, Hawtin R, et al. Filgotinib, a JAK1 Inhibitor. Rheumatol Ther. 2020;7(1):173–90.
38. Harigai M, Honda S. Selectivity of Janus kinase inhibitors in rheumatoid arthritis and other immune-mediated inflammatory diseases: is expectation the root of all headache? Drugs 2020;80(12):1183–201.
39. Ho Lee Y, Gyu Song G. Comparative efficacy and safety of tofacitinib, baricitinib, upadacitinib, filgotinib and peficitinib as monotherapy for active rheumatoid arthritis. J Clin Pharm Ther. 2020;45(4):674–81.
40. Lee YH, Song GG. Comparative efficacy and safety of tofacitinib, baricitinib, upadacitinib, and filgotinib in active rheumatoid arthritis refractory to biologic disease-modifying antirheumatic drugs. Z Rheumatol 2020. DOI:10.1007/s00393-020-00796-1.
41. Lee YH, Song GG. Relative efficacy and safety of tofacitinib, baricitinib, upadacitinib, and filgotinib in comparison to adalimumab in patients with active rheumatoid arthritis. Z Rheumatol. 2020;79(8):785–96.
42. Song GG, Lee YH. Comparative efficacy and safety of 100 mg and 200 mg filgotinib administered to patients with active rheumatoid arthritis: a Bayesian network meta-analysis of randomized controlled trials. Int J Clin Pharmacol Ther. 2020;58(06):293–8.
43. Lee YH, Song GG. Comparison of the efficacy and safety of tofacitinib and filgotinib in patients with active rheumatoid arthritis: a Bayesian network meta-analysis of randomized controlled trials. Z Rheumatol. 2020;79(6):590–603.
44. Rogler G. Efficacy of JAK inhibitors in Crohn’s disease. J Crohns Colitis. 2020;14(2):S746–S54.
45. Duggan S, Keam SJ. Upadacitinib: first approval. Drugs 2019;79(16):1819–28.
46. Mease PJ, Lertratanakul A, Anderson JK, Papp K, Van den Bosch F, Tsuji S, et al. Upadacitinib for psoriatic arthritis refractory to biologics: SELECT-PsA 2. Ann Rheum Dis. 2021;80(3):312–20.
47. van der Heijde D, Song I-H, Pangan AL, Deodhar A, van den Bosch F, Maksymowych WP, et al. Efficacy and safety of upadacitinib in patients with active ankylosing spondylitis (SELECT-AXIS 1): a multicentre, randomised, double-blind, placebo-controlled, phase 2/3 trial. Lancet. 2019;394(10214):2108–17.
48. Gilead and Galapagos announce positive topline results of phase 2b/3 trial of filgotinib in moderately to severely active ulcerative colitis . 2020. [2020-10-07] Available from: https://www.businesswire.com/news/home/20200520005818/en/.
49. Mease P, Coates LC, Helliwell PS, Stanislavchuk M, Rychlewska-Hanczewska A, Dudek A, et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active psoriatic arthritis (EQUATOR): results from a randomised, placebo-controlled, phase 2 trial. Lancet 2018;392(10162):2367–77.
50. van der Heijde D, Baraliakos X, Gensler LS, Maksymowych WP, Tseluyko V, Nadashkevich O, et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active ankylosing spondylitis (TORTUGA): results from a randomised, placebo-controlled, phase 2 trial. Lancet 2018;392(10162):2378–87.
51. Pfizer, Inc. Xeljanz® (tofacitinib) prescribing information (Revised 05/2018). [2020-09-22] Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/203214s018lbl.pdf.


