Introduction
Graft-versus-host disease (GVHD) is a major barrier to the success of hematopoietic stem cell transplantation (HSCT), affecting both survival and quality of life. Macrophages play a dual role in GVHD—acting as both therapeutic tools and targets. In acute GVHD, macrophages mostly polarize into the M1 phenotype, releasing pro-inflammatory cytokines that cause tissue damage. In chronic GVHD, they shift toward the M2 phenotype, driving tissue repair and fibrosis. Their interactions with T cells, B cells, and fibroblasts further shape disease progression—promoting inflammation in acute GVHD and fibrosis in chronic GVHD.
Current challenges include incomplete understanding of cytokine-driven macrophage polarization and the lack of safe targeted therapies. Future directions lie in macrophage-modulating approaches—using small molecules, antibodies, and nanotechnology—to reduce GVHD severity and enhance HSCT success.
Mechanism of GVHD
Mechanism of GVHD
GVHD arises mainly from tissue damage due to chemotherapy/radiation and HLA mismatch, which triggers alloreactive T cells. In acute GVHD, antigen presentation, T-cell activation, and cytokine networks lead to apoptosis and inflammation. Chronic GVHD involves thymus damage, abnormal B-cell and macrophage activity, Th17/Tfh imbalance, Treg suppression, and fibrosis. Macrophages amplify both acute (via DAMP recognition and inflammation) and chronic (via PAMP response, sustained damage, fibrosis) GVHD.
Macrophage heterogeneity and polarization
Macrophages are highly adaptable immune cells. Naive M0 macrophages can polarize into M1 (pro-inflammatory) or M2 (anti-inflammatory, tissue-repairing) depending on microenvironmental signals. This plasticity drives their roles in infection, repair, fibrosis, and immune regulation.
M1 polarized macrophages
M1 macrophages are induced by LPS and Th1 cytokines (IL-12, IFN-γ, TNF-β). They produce pro-inflammatory cytokines (IL-1, IL-6, TNF-α), ROS/RNS, and present antigens via MHC II. Key roles include pathogen clearance, tissue damage response, antigen presentation, and initiating repair. Polarization is driven by STAT1, IRF5, NF-κB, and TLR signaling.
M2 polarized macrophages
M2 macrophages are anti-inflammatory, promoting wound healing, fibrosis, and immune regulation. Induced by Th2 cytokines (IL-4, IL-10, IL-13), they secrete IL-10, TGF-β, and support tissue repair but can also promote tumors.
- M2a: debris clearance, tissue repair (IL-4/IL-13 driven).
- M2b: regulatory, balancing inflammation via IL-10.
- M2c: deactivated, anti-inflammatory, high phagocytosis of apoptotic cells.
- M2d (TAMs): tumor-associated, promote angiogenesis and cancer progression.
Emerging tools like scRNA-seq show more subtypes, reinforcing that M1–M2 balance is key for homeostasis, with imbalance leading to inflammation, autoimmunity, or poor wound healing.
Immune cell crosstalk and macrophage polarization
Immune cells regulate macrophage polarization mainly through cytokine signaling. IFN-γ from Th1 cells, NK cells, and macrophages drives M1 polarization via JAK/STAT1, enhancing inflammation and cytotoxicity. In contrast, IL-4 and IL-13 from Th2 cells activate STAT6 and PPARγ pathways, pushing macrophages toward an M2, tissue-repairing phenotype. IL-10 from Tregs and B cells further supports M2 activation through STAT3 while suppressing pro-inflammatory signals.
MicroRNAs in macrophage polarization
MicroRNAs fine-tune macrophage fate. M1 polarization is strengthened by miR-9, miR-125b, and miR-155, which enhance IFN-γ signaling and NF-κB activation. In contrast, miR-124 promotes M2 by suppressing TNF-α and IL-6, while miR-223 limits NF-κB/JNK signaling and inflammatory cytokine release. Together, these miRNAs control the balance between pro-inflammatory and anti-inflammatory macrophage states.
Macrophage polarization in GVHD
Macrophage polarization influences both outcomes and pathology in GVHD. Acute GVHD shows dominant M1 activity, driving inflammation and tissue damage, and a high M1/M2 ratio predicts poor prognosis. In chronic GVHD, M2 macrophages (especially CD163+ subsets) dominate, contributing to fibrosis and organ dysfunction. Soluble CD163 also serves as a biomarker for chronic GVHD risk.
Macrophages in GVHD pathogenesis
Recruitment and activation of macrophages in GVHD
Chemokines guide macrophage recruitment and polarization during GVHD. CXCR3 promotes M1 activation, while CCR2/CCR4 favor M2. CXCL2 and MCP-1 are central in attracting macrophages and T cells to target tissues, with their inhibition reducing GVHD severity. In chronic GVHD, CCL9 enhances macrophage infiltration and fibrosis, while CCL15 may serve as a biomarker. Drugs like pirfenidone reduce macrophage migration and fibrosis by blocking chemokine-driven pathways.
Role of macrophages in tissue damage
Macrophages activated by DAMPs and PAMPs amplify GVHD by releasing inflammatory cytokines and stimulating donor T cells. This leads to tissue injury in skin, liver, gut, and hematopoietic systems. In chronic GVHD, donor-derived macrophages promote fibrosis through TGF-β and PDGF signaling, driving fibroblast activation and collagen deposition. Therapies like pirfenidone and Axatilimab reduce macrophage infiltration and profibrotic cytokines. Excess IL-6 and IL-17 further worsen inflammation, favoring Th17 over Tregs, thereby sustaining cGVHD.
Macrophages and cytokines
Cytokines from macrophages and APCs initiate and shape GVHD progression. PAMPs and DAMPs activate TLRs, triggering IL-6, IL-12, and IL-23 release, which drive Th1/Th17 responses. M1 polarization is enhanced by metabolites like TMAO, increasing IL-1β, IL-6, and TNF-α. In chronic GVHD, macrophage-derived TGF-β and M-CSF promote fibrosis. Transcriptional profiling shows acute GVHD macrophages have a remodeling signature, while chronic GVHD macrophages are more pro-inflammatory. Overall, TNF-α, IL-6, and IL-12 dominate in acute GVHD, whereas TGF-β defines chronic GVHD.
Communication between macrophages and other immune cells
Macrophages influence T-cell activation and differentiation through cytokine release, antigen presentation, and polarization. By increasing IL-6, IL-1β, and IL-23, they drive Th17 polarization, worsening lung GVHD. Reduced macrophage infiltration shifts T-cell balance toward Th2/Treg, reducing aGVHD. Macrophages also interact with B cells—either promoting fibrosis by IL-6/TGF-β-producing subsets or suppressing antibody production. Thus, macrophages regulate both effector and regulatory immune responses, shaping GVHD severity.
Macrophage metabolism in GVHD
M1 macrophages rely on glycolysis, producing intermediates like citrate, succinate, and itaconate that drive ROS, cytokine release, and antimicrobial activity. HIF1α promotes glycolysis and lactate production, fueling inflammation. In contrast, M2 macrophages depend on oxidative phosphorylation and fatty acid oxidation, with α-ketoglutarate supporting anti-inflammatory function by suppressing HIF1α. This metabolic reprogramming defines their pro- or anti-inflammatory roles in GVHD.
Macrophages and GVHD-related diseases
Macrophages are central to GVHD pathogenesis, interacting with DAMPs, PAMPs, and alloreactive T cells to amplify inflammation and complications (Fig. 2). M1 macrophages dominate in acute GVHD, driving cytokine storms and tissue damage, while M2 macrophages prevail in chronic GVHD, promoting fibrosis via TGF-β. Distinct gene profiles link GVHD macrophages to other inflammatory diseases, such as ulcerative colitis. Elevated calprotectin+ macrophages in acute GVHD and gut microbiota–driven TH1/TH17 responses further highlight their role as biomarkers and mediators of disease progression.
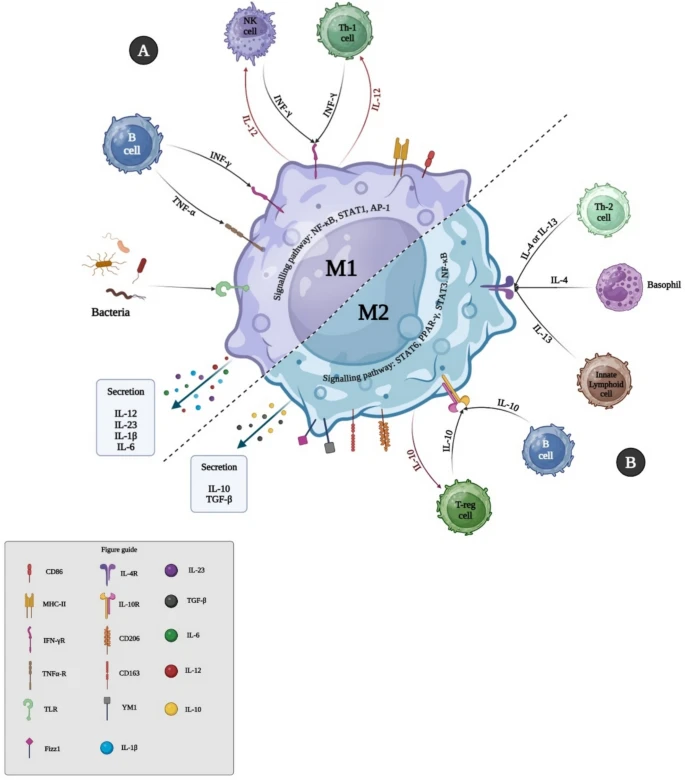
Figure 2.
Macrophages play a central role in both acute and chronic GVHD, shaping inflammation, tissue damage, and fibrosis. In acute GVHD, M1 macrophages dominate, producing proinflammatory cytokines that drive tissue injury, while chronic GVHD is linked to M2 macrophages, which promote tissue repair but also fibrosis. Their infiltration into skin, gut, liver, lungs, and bone marrow strongly correlates with GVHD onset and severity, with CD163+ M2 macrophages even serving as predictors of resistant and chronic disease. Tissue-resident and monocyte-derived macrophages further influence disease progression, with donor-derived populations aggravating keratinocyte and gut injury.
The balance between M1 and M2 states is critical but highly dynamic, shaped by cytokine networks and interactions with T cells, B cells, and fibroblasts. Th17 cytokines like IL-17 can dampen macrophage infiltration and inflammation, while therapies such as ROCK2 inhibitors or pirfenidone aim to restore immune tolerance, reduce fibrosis, and regulate macrophage activity. Pharmacological agents that block proinflammatory macrophages or promote M2 polarization are being explored to improve GVHD management.
Beyond direct cell activity, macrophage-derived extracellular vesicles (EVs) also regulate GVHD. M1-derived EVs enhance T-cell activation and inflammation via cytokines and miRNAs such as miR-155, while others like miR-223 and miR-124 promote anti-inflammatory effects and tissue protection. Together, these findings highlight macrophages as both mediators of GVHD pathology and promising therapeutic targets, with potential strategies ranging from cytokine modulation to macrophage-targeted therapies and EV-based interventions.
The role of the microbiome in shaping macrophage function in GVHD
The human gut hosts a diverse microbiota composed mainly of Firmicutes, Bacteroidetes, Actinomycetes, and Proteobacteria, which play essential roles in nutrient absorption, metabolism, immune defense, and the production of bioactive metabolites such as SCFAs, bile acids, and tryptophan derivatives. These metabolites regulate host physiology, including digestion, metabolism, and immune responses.
In allo-HSCT, conditioning regimens and antibiotics disturb gut microbial balance, leading to reduced microbial diversity, intestinal barrier dysfunction, and increased GVHD risk. Early studies showed that germ-free mice or those treated with antibiotics had reduced GVHD, while later clinical studies confirmed that loss of microbial diversity after HSCT is linked to higher GVHD incidence, severity, and mortality. Specific shifts—such as decreases in Clostridium, Faecalibacterium, and Bacteroides and increases in Enterococcus and Akkermansia—are associated with GVHD development.
Gut microbiota also shape immune balance by influencing Treg/Th17 and Th1/Treg ratios. Dysbiosis promotes proinflammatory pathways and fibrosis, while beneficial metabolites like SCFAs and tryptophan derivatives help maintain tolerance and protect intestinal integrity. Altering microbiota composition toward butyrate producers, or leveraging microbial metabolites, has shown promise in mitigating GVHD and represents a potential therapeutic strategy, though the underlying mechanisms remain incompletely understood.
Conclusion
This review highlights the crucial role of macrophages in GVHD and their interactions with the disease microenvironment. Targeting macrophage polarization and developing advanced delivery strategies hold promise for new therapies. However, addressing safety, refining targeting, and validating through clinical trials are essential. Successful translation of macrophage-based therapies could greatly improve GVHD management and patient outcomes after stem cell transplantation.
References
1. Li X, Gao Q, Feng Y, Zhang X. Developing role of B cells in the pathogenesis and treatment of chronic GVHD. Br J Haematol. 2019;184(3):323–36.
2. Saidu NEB, Bonini C, Dickinson A, Grce M, Inngjerdingen M, Koehl U, et al. New approaches for the treatment of chronic graft-versus-host disease: current status and future directions. Front Immunol. 2020;11:578314.
3. Michniacki TF, Choi SW, Peltier DC. Immune suppression in allogeneic hematopoietic stem cell transplantation. Handb Exp Pharmacol. 2022;272:209–43.
4. Aghaei M, Khademi R, Bahreiny SS, Saki N. The need to establish and recognize the field of clinical laboratory science (CLS) as an essential field in advancing clinical goals. Health Sci Rep. 2024;7(8):e70008.
5. Porrata LF. Autologous graft-versus-tumor effect: reality or fiction? Adv Hematol. 2016;2016:5385972.
6. Zeiser R. Introduction to a review series on pathophysiology and treatment of acute GVHD. Blood. 2020;136(4):375–6.
7. Socie G, Michonneau D. Milestones in acute GVHD pathophysiology. Front Immunol. 2022;13:1079708.
8. Teshima T, Hill GR. The pathophysiology and treatment of graft-versus-host disease: lessons learnt from animal models. Front Immunol. 2021;12:715424.
9. Ghimire S, Weber D, Mavin E, Wang XN, Dickinson AM, Holler E. Pathophysiology of GVHD and other HSCT-related major complications. Front Immunol. 2017;8:79.
10. Yu J, Hamilton BK, Turnbull J, Stewart SK, Vernaya A, Bhatt V, et al. Patient-reported symptom burden and impact on daily activities in chronic graft-versus-host disease. Cancer Med. 2023;12(3):3623–33.