Introduction
High-throughput technologies have revolutionized the understanding of rheumatoid arthritis (RA), revealing new molecular and cellular pathways in tissue inflammation. Fibroblasts, long studied in RA, have gained renewed attention as research identifies their role in immune responses, tissue remodeling, and treatment resistance. Specific fibroblast subpopulations have been characterized in both human and animal models, reshaping their perceived impact on disease progression and clinical relapses.
The lung is a crucial non-synovial site in RA, with interstitial lung disease (ILD) contributing significantly to morbidity and mortality. Despite advancements in ILD research, gaps remain in understanding its pathogenesis and optimizing diagnosis and treatment. While inflammation and fibrosis are recognized hallmarks of RA-ILD, the precise contribution of fibroblasts to lung involvement remains unclear, highlighting the need for further investigation.
Recent studies using high-throughput RNA sequencing have identified distinct fibroblast subpopulations in RA synovium, yet their role in RA-ILD remains underexplored. Fibroblasts play key roles in lung development, injury response, and repair, but their involvement in RA-associated lung disease is not well defined. This review delves into fibroblast-related genetic and immune mechanisms in RA-ILD, exploring their influence on disease evolution, flares, and treatment outcomes in both human and experimental models.
Overview of RA pathogenesis
The discovery of anti-citrullinated protein antibodies (ACPAs) has significantly advanced the understanding of RA pathogenesis. ACPAs, present in 50–60% of RA patients, are highly specific for the disease, while rheumatoid factors (RF) have lower specificity. RA is believed to develop over years, with early immune responses targeting post-translationally modified proteins in mucosal areas before spreading to synovial tissue. This leads to chronic synovitis, driven by inflammatory amplification and stromal cell transformation. (Fig. 1)
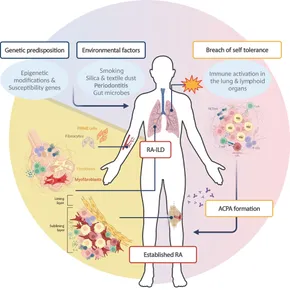
Figure 1. Rheumatoid arthritis pathogenesis: specific contribution of fibroblasts in RA phenotype.
Genetic and environmental factors play key roles in RA development. HLA-DRβ1 alleles, particularly those with a shared epitope, increase disease risk by enhancing T-cell activation via citrullinated peptides. Genome-wide association studies (GWAS) have identified over 100 additional risk alleles linked to immune pathways. Smoking, a strong environmental trigger, acts as an epigenetic modifier, promoting ACPA formation and autoimmune responses in the lung, which later contribute to joint inflammation. Impaired T-cell tolerance, metabolic alterations, and premature aging further drive tissue invasion and inflammation.
High-throughput technologies, such as single-cell RNA sequencing, have identified distinct synovial fibroblast, macrophage, T-cell, and B-cell subpopulations that contribute to chronic inflammation and joint damage. These findings have improved the understanding of RA’s cellular landscape, but further mechanistic studies are needed to determine how these immune and stromal cell subsets influence disease progression and treatment response.
Synovial fibroblasts in RA pathogenesis
Fibroblasts play a key role in RA by transforming from normal joint-supporting cells into aggressive, hyperactivated cells that degrade cartilage and bone. These fibroblast-like synoviocytes (FLS) promote inflammation, facilitate immune cell infiltration, and worsen disease progression. Advanced research has identified distinct fibroblast subtypes affecting prognosis and treatment response. Some fibroblast-rich patients show multidrug resistance, while others respond better to specific therapies. Studies also reveal fibroblast migration spreading RA between joints, highlighting their impact on disease flares and progression.
Synovial fibroblast heterogeneity
RA fibroblasts exhibit significant heterogeneity, with distinct subsets driving inflammation and tissue destruction. Single-cell studies in mice and humans identify two key fibroblast types: FAPα+THY1+ (inflammatory) and FAPα+THY1– (destructive). Their depletion reduces arthritis, while their transfer worsens disease. Spatial transcriptomics further reveals fibroblast variation in RA patients, linking specific subsets to macrophage-rich areas and plasma cells. Synovial fibroblasts in RA show increased invasiveness and inflammatory signaling, with TNF, IFN-γ, and IL-1β influencing their heterogeneity. Notably, RA fibroblasts differ from those in spondylarthritis, which exhibit a more fibrotic profile.
The lung as the site of initial autoimmune responses in RA
Rheumatoid Arthritis (RA) may originate outside the joints, with the lung playing a key role in triggering autoimmunity. Smoking, a major environmental risk factor, induces protein citrullination, leading to Anti-Citrullinated Protein Antibody (ACPA) formation, especially in Human Leukocyte Antigen-DR4 (HLA-DR4) carriers. Pulmonary involvement, including Interstitial Lung Disease (ILD) and airway abnormalities, appears early in RA. ACPA-positive RA patients show more lung changes than ACPA-negative ones. Studies suggest smoking and environmental exposures like silica and textile dust increase ACPA-positive RA risk. Elevated Peptidylarginine Deiminase 2 (PAD2) expression, immune activation, and Neutrophil Extracellular Trap Formation (NETosis) in the lungs support the hypothesis that RA-related autoimmunity may begin in the lung before joint inflammation develops.
Pathophysiology of RA-associated ILD
Pulmonary complications are the most common extra-articular manifestations in Rheumatoid Arthritis (RA), affecting up to 15% of patients clinically and up to 60% subclinically. Interstitial Lung Disease (ILD) is the most frequent RA-associated lung disease, impacting lung function and increasing morbidity and mortality. ILD often develops early and may precede joint symptoms. Usual Interstitial Pneumonia (UIP) is the most severe subtype, linked to older age, male gender, and smoking history, while Non-Specific Interstitial Pneumonia (NSIP) is more common in women.
Genetic (MUC5B, HLA genes), environmental (smoking), and clinical factors (high Anti-Citrullinated Protein Antibody (ACPA) and Rheumatoid Factor (RF) levels) contribute to RA-ILD risk. Some RA-ILD patterns share similarities with Idiopathic Pulmonary Fibrosis (IPF), suggesting common fibrotic pathways. Fibrocytes, immune cells, and inflammatory mediators like Interleukin-17A (IL-17A) play key roles in disease progression. Anti-fibrotic drugs such as pirfenidone and nintedanib may slow disease progression, particularly in UIP cases, but further research is needed to determine their efficacy as first-line treatments. Understanding fibroblast involvement could help develop targeted therapies for both lung and joint inflammation in RA.
Fibroblasts in lung fibrosis
Mesenchymal cells in the lung, located between the epithelium and stroma, play a crucial role in development and injury repair. Following injury, lung mesenchyme, including various fibroblast subsets, balances epithelial repair and pathological remodeling, sometimes leading to fibrosis. Lipofibroblasts (LIFs) can transform into profibrotic myofibroblasts due to nicotine, hyperoxia, or bleomycin exposure, driven by TGF-β1 signaling. Fibroblasts from IPF patients show increased invasiveness linked to HAS2, CD44, and HER2 signaling, with HER2 inhibition improving fibrosis when combined with anti-PDL1 treatment.
Single-cell RNA sequencing of lung biopsies from SSc-ILD patients identified expanded ACTA2-expressing myofibroblasts with high collagen production. Myofibroblasts, characterized by αSMA expression, enhance migratory and contractile abilities and are involved in various diseases, including cancer, fibrosis, and wound healing. Mechanical loading and TGF-β signaling regulate their activation, making them a target for RA therapies.
Animal models of arthritis show limited lung fibrosis, resembling NSIP more than UIP. Some models, like SKG and adjuvant-induced arthritis rats, exhibit mixed inflammatory and fibrotic lung phenotypes. The K/BxN model shows ectopic lymphoid tissue in the lung but lacks fibrosis progression. While collagen-induced arthritis models mimic RA-ILD, pulmonary lesions in animals tend to regress, unlike in humans. More refined models are needed to study RA-ILD pathogenesis accurately.
Therapeutic targeting of fibroblasts
Fibroblasts play a major role in RA-related inflammation and joint damage but are not directly targeted by advanced treatments like anti-CD20 (B-cells) and CTLA4-Ig (T-cells). While TNF inhibitors and IL-6 blockers may impact fibroblast activity, studies suggest TNF/TNFR signaling via the p55TNFR–IKK2–Ripk3 axis is crucial for fibroblast survival and inflammation, making Ikk2 a potential therapeutic target.
Jak inhibition (upadacitinib) has been shown to reverse the activation of RA synovial fibroblasts, unlike TNF inhibitors. In RA-ILD models, the Jak inhibitor baricitinib reduced fibrosis by blocking TGF-β1/non-Smad and Jak/STAT pathways, limiting fibroblast activation and epithelial damage. It also attenuated pulmonary fibrosis in collagen-induced arthritis by inhibiting Jak2/Stat3 signaling.
Conclusion
Fibroblasts play a crucial role in RA pathogenesis, progression, and treatment response, both in the synovium and the lung. Their aggressive states are linked to severe disease and poor treatment outcomes. While studies in animal models and cell cultures have provided insights into fibroblast-targeting therapies, their clinical application remains limited.
Further research is needed to understand fibroblast functions in RA, especially in RA-ILD, where molecular mechanisms remain unclear. Collaborative efforts are essential to map fibroblast interactions, identify biomarkers, and translate findings into clinical practice.
References
1. Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. (2002) 347:911–20. doi: 10.1056/NEJMra020100
2. Cross M, Smith E, Hoy D, Carmona L, Wolfe F, Vos T, et al. The global burden of rheumatoid arthritis: Estimates from the Global Burden of Disease 2010 study. Ann Rheum Dis. (2014) 73:1316–22. doi: 10.1136/annrheumdis-2013-204627
3. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
4. Winthrop KL, Mease P, Kerschbaumer A, Voll RE, Breedveld FC, Smolen JS, et al. Unmet need in rheumatology: Reports from the Advances in Targeted Therapies meeting, 2023. Ann Rheum Dis. (2023) 83:409–16. doi: 10.1136/ard-2023-224916
5. Poppelaars PB, van Tuyl LHD, Boers M. Normal mortality of the COBRA early rheumatoid arthritis trial cohort after 23 years of follow-up. Ann Rheum Dis. (2019) 78:586–9. doi: 10.1136/annrheumdis-2018-214618
6. Zhang Y, Lu N, Peloquin C, Dubreuil M, Neogi T, Aviña-Zubieta JA, et al. Improved survival in rheumatoid arthritis: A general population-based cohort study. Ann Rheum Dis. (2017) 76:408–13. doi: 10.1136/annrheumdis-2015-209058
7. Holmqvist M, Ljung L, Askling J. Mortality following new-onset Rheumatoid Arthritis: Has modern Rheumatology had an impact? Ann Rheum Dis. (2018) 77:85–91. doi: 10.1136/annrheumdis-2017-212131
8. Gabbay E, Tarala R, Will R, Carroll G, Adler B, Cameron D, et al. Interstitial lung disease in recent onset rheumatoid arthritis. Am J Respir Crit Care Med. (1997) 156(2 Pt. 1):528–35. doi: 10.1164/ajrccm.156.2.9609016
9. Koliaraki V, Prados A, Armaka M, Kollias G. The mesenchymal context in inflammation, immunity and cancer. Nat Immunol. (2020) 21:974–82. doi: 10.1038/s41590-020-0741-2
10. Zhang F, Jonsson AH, Nathan A, Millard N, Curtis M, Xiao Q, et al. Deconstruction of rheumatoid arthritis synovium defines inflammatory subtypes. Nature. (2023) 623:616–24.