Introduction
Cardiovascular disease (CVD) is a leading cause of mortality among chronic renal disease patients (CKD). Those with poor renal function have a 40–50 percent higher risk of developing coronary artery disease (CAD) than patients with adequate renal function, according to large meta-analyses.
This may be mediated, at least in part, by the fact that hypertension and diabetes mellitus, two of the most frequent causes of CKD, have also been recognized as cardiovascular risk factors. According to new research, CKD is an independent risk factor for CVD. CKD-specific changes that enhance cardiovascular risk include the accumulation of uremic toxins, chronic inflammation, and oxidative stress (Table 1)

Vascular changes, particularly atherosclerosis and vascular calcification, have a significant impact on the link between CKD and cardiovascular mortality (VC). VC is the primary mechanism through which CKD affects the medial layer of blood arteries. It is a multifactorial, cell-mediated process in which a variety of disorders, such as mineral dysregulation and hyperphosphatemia, cause a phenotypic flip in vascular smooth muscle cells to osteoblast-like cells.
Intercellular communication channels via extracellular vesicles and microRNAs are essential processes in VC, according to recent research, and thus a potential topic for a fuller knowledge of the associated pathomechanisms. In this review, we discuss the pathophysiological processes that link CKD and CVD, as well as newly identified molecular pathways that might be used as novel therapeutic targets.
Pathophysiology and therapeutic targets
Chronic inflammation
Atherosclerosis and its manifestation in the coronary arteries, CAD, is supposed to be a key connector between CKD and cardiovascular morbidity and mortality.
Elevated levels of inflammatory biomarkers such as C-reactive protein (CRP), interleukin-6 (IL-6), or tumor necrosis factor (TNF) have been shown to be associated with an increased risk of myocardial infarction and mortality. Recent controlled randomized clinical trials have demonstrated a beneficial effect of SGLT2-inhibitors in heart failure patients with and without diabetes regarding the occurrence of major cardiovascular endpoints.
Of note, SGLT2-inhibition was additionally associated with a slower progression of CKD in patients with and without diabetes. Aside from an improved glycemic control, SGLT2-inhibitors is widely focused on volume control via the induction of natriuresis and osmotic diuresis. Chronic inflammation in CKD is a multifactorial condition and specific pharmacological targets to improve outcomes for patients are rare. IL-1β inhibition has shown promising results in animal models with different renal disorders.
Vascular calcification (VC) in CKD
While atherosclerosis is characterized through vascular endothelial dysfunction progressing to vascular structural damage, the medial layer of blood vessels is affected differently in CKD. VC manifesting in the coronary arteries impairs coronary flow reserve and is associated with a increase in adverse cardiac events and cardiovascular mortality. VC occurs in two different phenotypes, medial and intimal calcification, differing in their
pathogenesis. While intimal calcification is mainly inflammation-driven and closely associated to atherosclerotic plaques, medial calcification is considered to be the major form of VC in CKD (Fig 1).

Meta-analyses of available randomized controlled trials regarding phosphate binding agents have demonstrated a lower all-cause mortality of patients receiving the non-calcium-based agent sevelamer compared to calcium-based binders while there was no significant difference regarding cardiovascular death.
In this nondialysis CKD population, the patients presented with normophosphatemia at baseline. Clinical data suggest an inverse association between serum magnesium levels and the expression of VC in peritoneal- and hemodialysis patients. More recently, impairment of calcification inhibiting mechanisms, such as fetuin A, klotho, and the vitamin K-dependent matrix
Endothelial dysfunction
Accumulating evidence suggests that CKD promotes atherosclerosis and CAD by inducing damage to endothelial cells. Albuminuria as a consequence of glomerular damage was shown in several studies to correlate
with elevated levels of von Willebrand factor, an indicator of endothelial dysfunction. Important mediators of endothelial dysfunction in CKD are uremic toxins such as asymmetric dimethylarginine (ADMA) and indoxyl sulfate (IS), which accuGla protein (MGP) and Gla-rich protein (GRP) have emerged as participants in the complex multifactorial process of VC. Another key calcification inhibitor which is impaired in CKD patients is the klotho/FGF23 axis.
Evidence suggests that klotho improves the phosphate metabolism by inducing phosphaturia and supresses thereby VSMC calcification. Vitamin K1 supplementation however is a promising and uncomplicated way to positively influence VC in CKD patients and, therefore, its effect on CV risk is under investigation in current clinical trials (Table 2).

accumulate in CKD patients in parallel with declining renal function. ADMA disturbs endothelial function by competitive inhibition of eNOS and is closely associated with the presence and functional significance of CAD in CKD. Another recently described mechanism of endothelial dysfunction in CKD is driven by a change in the functional properties of LDL-cholesterol. Hyperphosphatemia as a frequently observed finding among patients with CKD is known to be a major factor involved in the development of medial calcification in CKD.
Extracellular vesicles as intercellular messengers in CKD and CAD
In the past years, circulating EV have emerged to play a key role in cardiovascular health and disease. EV play an important role in intercellular communication by transferring their bioactive cargos including proteins, lipids, and nucleic acids. This mechanism of signal transfer has evolved as an important regulator of cardiovascular health and disease.
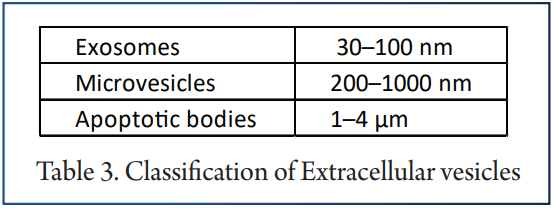
Circulating EV are known to mediate many physiological and pathophysiological mechanisms like inflammation and coagulation. Endothelial cell damage and apoptosis are crucial steps in the pathophysiology of CVD and endothelial microvesicles have been shown to be elevated in patients with CVD compared to healthy control subjects. Increased levels of endothelial EV are independent predictor of CV mortality in patients with ESRD.
In vitro, endothelial EV from plasma of patients with ESRD led to reduced endothelium-dependent relaxations and cyclic guanosine monophosphate (cGMP) generation. EV produced by VSMC under physiological conditions do not contain calcium phosphate crystals and moreover transport calcification inhibitory proteins such as vitamin K-dependent MGP and fetuin-A. Influencing the phenotype and cargo of EV derived from VSMC could be an interesting therapeutic target for VC in CKD patients.
MicroRNAs as gene regulators of vascular alteration in CKD
MicroRNAs (miRNAs) play an outstanding role in post transcriptional gene regulation and can be transferred intercellularly through EV. Packaging of endothelial miR-92a-3p into endothelial microvesicles regulates angiogenesis and may act as a potential regenerative messenger in intercellular communication.
Patients with diabetic nephropathy express a different exosomal miRNA profile than healthy subjects and upregulated miRNAs closely correlate with the degree of albuminuria. RUNX2, a transcription factor which regulates osteoblast differentiation and VSMC calcification, represents a key target for miRs influencing VC.
Recent data show protective miR like miR-133a, miR-204, and miR-205, as well as miR triggering VC like miR-32 influence RUNX2 by decreasing or inducing its expression. Furthermore, miR-30b and miR-125b have recently been identified to protect against VC. Bone morphogenetic protein-2 (BMP-2) may promote vascular calcification by decreasing miR-30b and miR-30c to induce RUNX2 expression whereas upregulation of miR-30b attenuated VC in vivo and in vitro.
MiR-125b levels correlated significantly with VC severity as well as levels of fetuin-A and mediators of mineral bone disorder like osteoprotegerin and FGF-23.
Summary
In conclusion, cardiorenal syndrome, which is a mix of cardiovascular and chronic kidney disease, is a major issue for modern medicine due to growing incidence and prevalence rates. Cardiovascular mortality is the primary cause of death among individuals with reduced renal function, and vascular calcification is a significant connection between CKD and cardiovascular mortality.
Although several possible pathophysiological processes have been discovered in recent years, many aspects remain unclear, and as a result, treatment choices for individuals with VC are limited. The development of novel treatment targets requires a better knowledge of the molecular mechanisms and genetic targets involved in the complicated process of VC.
Additional treatment targets with the potential for significant advances in therapy may be found in the future as a result of a better knowledge of the pathophysiological mechanisms involved in VC.
References
1. KDIGO Board Members (2013) KDIGO 2012 Clinical Practice Guideline for the Evaluation and Managment of Chronic Kidney Disease, Summary of Recommendation Statements. Kidney Int Suppl 3:5–14.
2. Thompson S, James M, Wiebe N et al (2015) Cause of death in patients with reduced kidney function. J Am Soc Nephrol 26:2504–2511
3. Matsushita K, van der Welde M, Astor BC et al (2010) Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet 375:2073–2081
4. Webster AC, Nagler EV, Morton RL, Masson P (2017) Chronic kidney disease. Lancet 389:1238–1252
5. Di Angelantonio E, Danesh J, Eiriksdottir G, Gudnason V (2007) Renal function and risk of coronary heart disease in general populations: new prospective study and systematic review. PLoS Med 4:e270.
6. Perkovic V, Verdon C, Ninomiya T, Barzi F, Cass A, Patel A, Jardine M, Gallagher M, Turnbull F, Chalmers J, Craig J, Huxley R (2008) The relationship between proteinuria and coronary risk: a systematic review and meta-analysis. PLoS Med 5:e207.
7. Tonelli M, Muntner P, Lloyd A et al (2012) Risk of coronary events in people with chronic kidney disease compared with those with diabetes: a population-level cohort study. Lancet 380:807–814
8. Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm LL, McCullough P, Kasiske BL, Kelepouris E, Klag MJ, Parfrey P, Pfeffer M, Raij L, Spinosa DJ, Wilson PW, American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention (2003) Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 108:2154–2169
9. Bae EH, Lim SY, Cho KH et al (2012) GFR and cardiovascular outcomes after acute myocardial infarction: results from the Korea Acute Myocardial Infarction Registry. Am J Kidney Dis 59:795–802
10. Nakano T, Ninomiya T, Sumiyoshi S, Fujii H, Doi Y, Hirakata H, Tsuruya K, Iida M, Kiyohara Y, Sueishi K (2010) Association of kidney function with coronary atherosclerosis and calcification in autopsy samples from Japanese elders: the Hisayama Study. Am J Kidney Dis 55:21–30
11. Shamseddin MK, Parfrey PS (2011) Sudden cardiac death in chronic kidney disease: epidemiology and prevention. Nat Rev Nephrol 7:145–154
12. Goodman WG, Goldin J, Kuizon BD et al (2000) Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med 342:1478–1483
13. London GM, Guérin AP, Marchais SJ et al (2003) Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant 18:1731–1740
14. Toussaint ND, Kerr PG (2007) Vascular calcification and arterial stiffness in chronic kidney disease: implications and management. Nephrology 12:500–509
15. Kim J, Bravo PE, Gholamrezanezhad A et al (2013) Coronary artery and thoracic aorta calcification is inversely related to coronary flow reserve as measured by 82Rb PET/CT in intermediate risk patients. J Nucl Cardiol 20:375–384
16. Raggi P (2017) Coronary artery calcification predicts risk of CVD in patients with CKD. Nat Rev Nephrol 13:324–326
17. Perkovic V, Hunt D, Griffin SV et al (2004) Accelerated progression of calcific aortic stenosis in dialysis patients. Nephron Clin Pract 94:c40–c45
18. Rattazzi M, Bertacco E, Del Vecchio A et al (2013) Aortic valve calcification in chronic kidney disease. Nephrol Dial Transplant 28:2968–2976
19. Patel KK, Shah SY, Arrigain S, Jolly S, Schold JD, Navaneethan SD, Griffin BP, Nally JV, Desai MY (2019) Characteristics and outcomes of patients with aortic stenosis and chronic kidney disease. J Am Heart Assoc 8:e009980
20. Tumlin JA, Costanzo MR, Chawla LS et al (2013) Cardiorenal syndrome type 4: insights on clinical presentation and pathophysiology from the Eleventh Consensus Conference of the Acute dialysis Quality Initiative (ADQI). In: McCullough PA, Kellum JA, Mehta RL et al (eds) Contributions to nephrology. S. KARGER AG Basel, pp 158–173
21. Oh J, Wunsch R, Turzer M, Bahner M, Raggi P, Querfeld U, Mehls O, Schaefer F (2002) Advanced coronary and carotid arteriopathy in young adults with childhood-onset chronic renal failure. Circulation 106:100–105